CLI Tools for DNA and RNA‐seq

Important
🕐 Schedule
- 3:00pm-3:05pm: Welcome and introdution to topic
- 3:05pm-3:10pm: The Command Line in <5 Minutes
- 3:10pm-3:20pm: Quality Control Tools
- 3:20pm-3:55pm: Sequence Alignment and Assembly Tools
- 3:55pm-4:00pm: Honorable Mentions Closing remarks
Important
❗ Requirements
- Basic command line knowledge
- Access to a Terminal
- Unix and Mac users already have access to the Terminal
- Windows users can use either PowerShell or the Windows Subsystem for Linux (WSL)
- A registered CyVerse account (Register for a CyVerse account)
Important
✅ Expected Outcomes
- Exposure to general bioinformatic tools
- Understanding of genome assembly pipelines
This workshop focuses on the various tools every bioinformatician should know about. This is NOT intended to be a deep dive into each and every tool, as each software is incredibly complex and requires precious time to truly be understood. However, this workshop tries to expose the most common tools when it comes to the following popular bioinformatic topics: quality control (QC), sequence alignment, transcript assembly, variant calling.
Jack of all trades, master of none, but oftentimes better than a master of one.
Warning
❓ "... but my favourite tool/topic is missing!"
That's completely understandable: bioinformatics/genomics is such a wide field; This list is non-exhaustive and I will add more tools you may be interested in at the end of the workshop's page.
❗ Please do notice that visualization tools and analysis software have their own sessions! Check the Data Science Institute's DataLab website for more information. ❗
Important
This workshop uses data obtained by the National Center of Biotechnology Information (NCBI).
We are going to be using E. coli as the example organism throughout this workshop. All files are made available on CyVerse here or if you are using the command line here: iplant/home/shared/cyverse_training/datalab/biosciences.
- The E. coli genome used will be the ASM584v2 (4.6MB).
- Curated RefSeq annotations (gff) for the genome can be found in the same location as above.
- Raw reads used are from here (302.8MB) and here (Note: these sequences come from the same project and have been processed with an Illumina NovaSeq 6000 machine).
- These reads have been fetched with the
sra-tools(prefetch <SRA sample>) and extracted (fastq-dump <SRA sample>.sra).
- These reads have been fetched with the
We are also going to be using some famous genes that access:
Most tools used in bioinformatics/genomics are command line tools (not taking into consideration Python or R tools).
The Command Line is how you communicate with your computer: it sends specific commands that tell software to execute specific actions. Following is a quick example of how commands work:
hostname/machine command input file argument/value----------------┐
↓ ↓ ↓ ↓ ↓ ↓
jovyan@aaf56ff92:~/data-store$ blastn -query <input.fasta> -db nt -out output.txt -evalue 1e-5
↑ ↑ ↑ ↑ ↑ ↑
user current location flag/option flags/options------------┘
\---------------------------/ \--------------------------------------------------------------/
shell prompt command
Note
- The shell prompt (or simply, prompt) is always followed by the command.
- The command can have a number of flag/options.
- Usually these can be listed by doing
<command> --help("help me by showing me your options!") - Each flag/option requires one of the following:
- The path to an input file;
- An argument (e.g., preferred name of output) or a value (e.g.,
1e-5) - Some options do not require any added argument/value (e.g.,
--helpdoes not require any further input)
- Usually these can be listed by doing
Tip
Always remember: Spaces in Paths are bad.
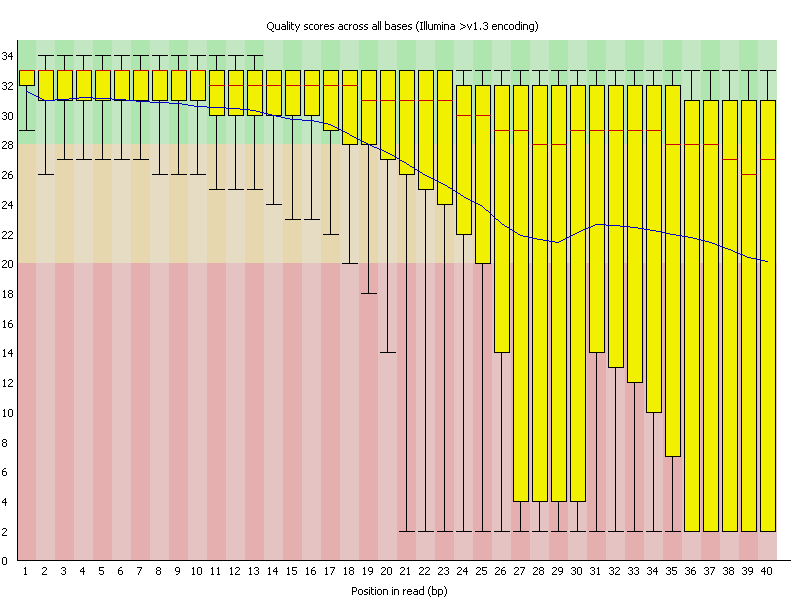
This section covers popular methods to assess quality of data. FastQC assesses raw data from sequencing runs, useful before an assembly or analysis starts. MultiQC takes outputs of other tools (but including FastQC) to give a more in depth report across multiple samples.
Upon obtaining raw sequencing data and before starting your downstream analysis, it is common that at the beginning of your pipeline you run FastQC.
FastQC will help you identify potential issues such as
- Low quality scores at certain positions.
- Adapter contamination.
- Overrepresented sequences.
- GC content anomalies.
The typical workflow including FastQC is the following:
- Run FastQC on the raw reads.
- Review the report for quality issues.
- Perform trimming/cleaning if needed (using tools like Trimmomatic or Cutadapt).
- Optionally, re-run FastQC to confirm the data is clean.
To run fastqc, the commands are
fastqc <sample_data>.fastq -o /path/to/output
Or, if you have multiple samples
fastqc *.fastq -o /path/to/output
Let's use the figure above to help us understand the output FastQC, called the the Per base sequence quality plot, and it provides the distribution of quality scores at each position in the read across all reads.
- The y-axis gives the quality scores, while the x-axis represents the position in the read.
- The color coding of the plot denotes what are considered high, medium and low quality scores.
- The yellow box represents the 25th and 75th percentiles, with the red line as the median. The whiskers are the 10th and 90th percentiles.
- The blue line represents the average quality score for the nucleotide.
Important
What's the Phred Quality Score?
The y-axis typically represents Phred quality scores, which measure the probability of an incorrect base call. Quality scores are logarithmic and can theoretically go beyond 40. Scores above 40 represent very high confidence in base calls, with probabilities of incorrect base calls being very low (less than 1 in 10,000). Setting the y-axis limit at 40 helps in standardizing the plot and avoiding unnecessary scaling for the majority of sequencing data, where scores often peak around 30 to 40. Scores higher than 40 are relatively rare in most datasets.

Other important results:
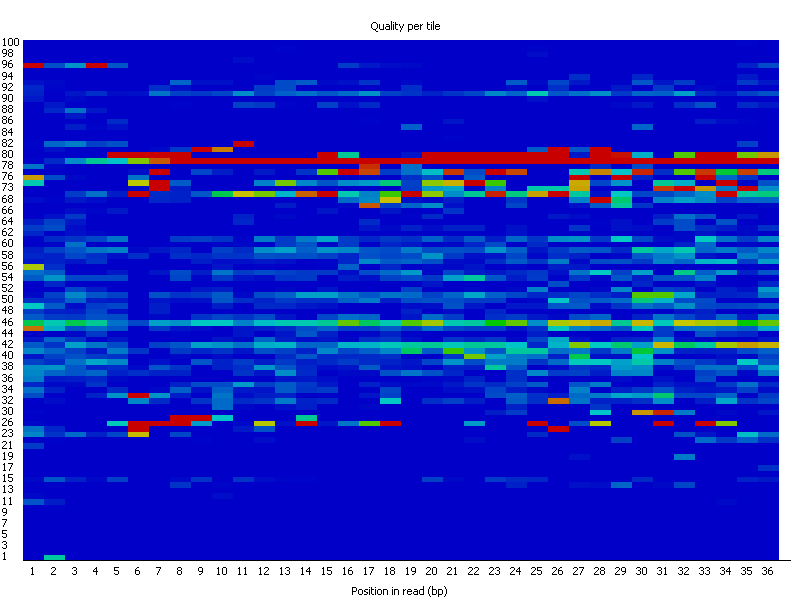
- Per tile sequence quality (only if sequencing was carried out with Illumina): The plot shows the deviation from the average quality for each tile, blue = good, red = bad (heat scale blue to red). Here's an example of a not so great result (source).
- Per sequence quality scores: the average quality score on the x-axis and the number of sequences with that average on the y-axis.
- Per base sequence content: plots out the proportion of each base position in a file for which each of the four normal DNA bases has been called. In a random library you would expect that there would be little to no difference between the different bases of a sequence run, so the lines in this plot should run parallel with each other. Note: this will always fail with RNA-seq data.
- Per sequence GC content: plots the GC distribution over all sequences
- Per base N content: If a sequencer is unable to make a base call with sufficient confidence then it will normally substitute an N rather than a conventional base call. This module plots out the percentage of base calls at each position for which an N was called.
- Sequence Length Distribution: shows the distribution of sequence length.
{kind=link}
MultiQC is similar to FastQC in a sense that both generate reports. However, MultiQC is able to aggregate reports from various sources and for various reasons including but not limited to:
- Sequence Quality Control (from FastQC)
- Alignment and Mapping (from STAR, HISAT2, Bowtiw2, BWA, Minimap2)
- Variant Calling (from GATK, Bcftools)
- Quantifiation (from Salmon, Kallisto)
- Differential Expression (DESeq2)
Notice how MultiQC relies on these tools to create reports first. As an aggregator, the power of MultQC lies in the ability to take all of your produced reports and have them all in a single place.
To run MultiQC, one must first designate a folder with all of the generated reports and analyses. Once the folder is created, once can generate the MultiQC report by doing
multiqc /path/to/folder
A report is going to be generated, similar to one available at in the data commons or data/iplant/home/shared/cyverse_training/datalab/biosciences/fastqc_multiqc_reports/ if you are following the workshop. Once logged in CyVerse, you should be able to view the report from your browser.
This MultiQC report was created by first creating the FastQC reports for samples SRR30597479 and SRR30597506. No other tool was aggregated to the report.
Image above was generated by the alignment software ClustalO, a powerful multiple sequence aligner (MSA) that allows for manual manipulation of each base.
Alignment tools are key in the realm of bioinformatics as these tools help with indefying regions of intetrest, similarites between regions, assemblies, and more for DNA, RNA or protein sequences. There's an acual plethora of aligners you have access to..
In this section, we are going to cover BLAST (Basic Local Alignment Search Tool), BWA (Burrows-Wheeler Aligner), Bowtie2, and HiSAT2
One of the most well known tools in the realm of bioinformatics is BLAST. It's one of the tools that is introduced very early on when learning bioinformatics, and is a staple for quick alignment searches. NCBI-BLAST is available and launchable from any browser and uses the NCBI compute in order to carry out analyses.
BLAST is able to search for both nucleotide sequences and protein sequences in a quick manner, making it an adequate tool for introducing bioinformatics to a newer audience.
On the command line, BLAST is an even more powerful tool, however it comes with a few too many options. Executing BLAST first requires a genome that has been made into a database with makeblastdb.
The full command used is makeblastdb -in ASM584v2.fna -dbtype nucl -out e_coli_db.
For this BLAST exercise, first copy the genome, annotations database and genes to BLAST onto your current location:
cp data/iplant/home/shared/cyverse_training/datalab/biosciences/e_coli_gen_dir/ data/iplant/home/shared/cyverse_training/datalab/biosciences/genes .
Then, you're ready to BLAST! The command to use is:
blastn -query <gene>.fa -db e_coli_gen_dir/e_coli_db/e_coli_db -out results.out
Important
What's going on here? Breaking down the command
-
blastn: calls the nucleotide version of BLAST -
-query <gene.fa>: here you point to the FASTA file to query the search -
-db e_coli_gen_dir/e_coli_db/e_coli_db: this points to where the E. coli database lives. Note: inside thee_coli_gen_dir/e_coli_dblives a number of files which end with a number of extensions; All together these files make up the database, callable withe_coli_db. -
-out results.out: creates an output file calledresults.out
To view the output, one can do:
cat results.out
If, for example, you blasted any other gene other than the lacZ gene, the results.out output will be:
BLASTN 2.16.0+
Reference: Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb
Miller (2000), "A greedy algorithm for aligning DNA sequences", J
Comput Biol 2000; 7(1-2):203-14.
Database: ASM584v2.fna
4,315 sequences; 4,025,874 total letters
Query= random_gene
Length=27
***** No hits found *****
Lambda K H
1.33 0.621 1.12
Gapped
Lambda K H
1.28 0.460 0.850
Effective search space used: 39525190
Database: ASM584v2.fna
Posted date: Sep 10, 2024 1:42 AM
Number of letters in database: 4,025,874
Number of sequences in database: 4,315
Matrix: blastn matrix 1 -2
Gap Penalties: Existence: 0, Extension: 2.5
However, running the lacZ gene, will result with:
BLASTN 2.16.0+
Reference: Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb
Miller (2000), "A greedy algorithm for aligning DNA sequences", J
Comput Biol 2000; 7(1-2):203-14.
Database: ASM584v2.fna
4,315 sequences; 4,025,874 total letters
Query= NC_000913.3:c366305-363231 Escherichia coli str. K-12 substr.
MG1655, complete genome
Length=3075
Score E
Sequences producing significant alignments: (Bits) Value
lcl|NC_000913.3_cds_NP_414878.1_338 [gene=lacZ] [locus_tag=b0344]... 5679 0.0
>lcl|NC_000913.3_cds_NP_414878.1_338 [gene=lacZ] [locus_tag=b0344]
[db_xref=UniProtKB/Swiss-Prot:P00722] [protein=beta-galactosidase]
[protein_id=NP_414878.1] [location=complement(363231..366305)]
[gbkey=CDS]
Length=3075
Score = 5679 bits (3075), Expect = 0.0
Identities = 3075/3075 (100%), Gaps = 0/3075 (0%)
Strand=Plus/Plus
Query 1 ATGACCATGATTACGGATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCT 60
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 1 ATGACCATGATTACGGATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCT 60
Query 61 GGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGC 120
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 61 GGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGC 120
Query 121 GAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGC 180
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 121 GAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGC 180
Query 181 TTTGCCTGGTTTCCGGCACCAGAAGCGGTGCCGGAAAGCTGGCTGGAGTGCGATCTTCCT 240
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 181 TTTGCCTGGTTTCCGGCACCAGAAGCGGTGCCGGAAAGCTGGCTGGAGTGCGATCTTCCT 240
Query 241 GAGGCCGATACTGTCGTCGTCCCCTCAAACTGGCAGATGCACGGTTACGATGCGCCCATC 300
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 241 GAGGCCGATACTGTCGTCGTCCCCTCAAACTGGCAGATGCACGGTTACGATGCGCCCATC 300
...............................................................................
..........................TRUNCATED TO SAVE SPACE..............................
...............................................................................
Lambda K H
1.33 0.621 1.12
Gapped
Lambda K H
1.28 0.460 0.850
Effective search space used: 11966980014
Database: ASM584v2.fna
Posted date: Sep 10, 2024 1:42 AM
Number of letters in database: 4,025,874
Number of sequences in database: 4,315
Matrix: blastn matrix 1 -2
Gap Penalties: Existence: 0, Extension: 2.5
Resulting in a successful BLAST hit!
There are more ways to customize your BLAST search. Other options include:
-
-outfmt: which allows you to pick the output format. Ther are 13 different output formats, tabular (-outfmt 6) is the most popular. -
-evalue: allows you to pick the E-value threshold (expected value), the number of hits you would expect to see by chance. Lower the E-value the more significant the match. A standard E-value is-evalue 1e-5 -
-max_target_seqs: limits the number of alignments to report per query sequence. E.g.,-max_target_seqs 10will report a max of 10 matches per sequence. -
num_threads: specifies the number of threads for parallel processing. A trick: a thread per computer core.
Note
As you may have noticed, BLAST is used to search for alignments between sequences, giving you a number of information per alignment. The following alignment tools are used for a different type of alignment: mapping reads and sequences in order to assemble genomes and RNA-seq data.
Important
Due to time constraints, we won't be able to play around with these. From here on, the workshop is going to be more theorical. We will be able to use some of these tools in the coming workshop dates. Note: not all flags/options are going to be covered in the next section as per each tool the complexity is extremely high.
BWA (Burrows-Wheeler Aligner) is a widely used software package for mapping sequencing reads against a reference genome. It is designed for high-throughput sequence alignment and is known for its speed and accuracy. BWA can handle both short and long reads and is commonly used in genomics for aligning reads from DNA sequencing experiments.
It has 3 algorithms:
- BWA-MEM: The most commonly used algorithm, especially for long reads and high-throughput sequencing. It is designed for accurate and fast alignment of reads.
- WA-ALN: Used for aligning short reads, particularly useful for older versions of sequencing technologies.
- BWA-SW: Used for aligning longer reads and for more sensitive alignment, but it is slower compared to BWA-MEM.
BWA use cases include:
- Variant Calling: Aligning sequencing reads to detect genetic variations such as SNPs and indels.
- Gene Expression Analysis: Aligning RNA-seq reads to measure gene expression levels.
- Structural Variation Detection: Identifying larger genomic rearrangements and structural variants.
The implementation of BWA in a pipeline is similar to the following:
- Index the Reference Genone
bwa index reference_genome.fasta
- Aligning Reads with BWA-MEM and redirect the output to a SAM file.
bwa mem reference_genome.fasta reads.fq > aligned_reads.sam
Note: reads.fq in the example above is a reads in a FASTQ format, a popular format that not only contains the sequence information, but also the quality score (similar to the Phred score discussed earlier -- the higher the better.) Quality scores are represented by a mix of symbols and letters (reference).
The output SAM file is a format used to store alignment data. SAM is human readable, whilst BAM is the computer readable counterpart (both can be outputted).
Important
As BWA is used mainly for shorter reads, a sister tool has been created in order to align longer reads (e.g., nanopore and PacBio sequences): Minimap2. The main difference is in the algorithms used, as Minimap2's algorithm is more efficient and accurate with longer read sequences. The two operate with a similar synthax.
A similar tool to BWA is Bowtie2, which is quicker, more memory efficient and can accomodate longer reads as well as short ones. The algorithms used are similar, with Bowtie2 also implemeting similar approaches to alignments that Minimap2 uses.
Bowtie2 is more ideal for small RNA-Seq experiments, whist BWA more useful for larger genome experiments.
Like BLAST and BWA, Bowtie2 first requires an indexed genome, using a sub tool called bowtie2-build:
bowtie2-build reference.fasta index_base_name
The options here are:
-
reference.fasta: The FASTA file containing the reference genome sequence. -
index_base_name: The base name for the index files that Bowtie2 will generate.
Upon indexing, you can then align:
bowtie2 -x index_base_name -1 reads_1.fastq -2 reads_2.fastq -S output.sam
The options are:
-
-x: Reference index. -
-1and -2: Paired-end reads (generated by the sequencer). -
-S: Output SAM file.
For alignning multiple paired-end reads using Bowtie2, you can concatenate them by separating them with a comma (,)
bowtie2 -x reference_index -1 reads_1a.fastq,reads_1b.fastq -2 reads_2a.fastq,reads_2b.fastq -S output.sam
STAR (Spliced Transcripts Alignment to a Reference) is a powerful tool that is specifically designed around RNA-seq experiments. It can align spliced reads, meaning it can handle the fact that RNA-Seq reads might span exons and introns. STAR’s ability to map reads across splicing junctions makes it ideal for analyzing transcriptomes, where splicing patterns need to be captured.
The downside of STAR is that it requires a lot more resources (RAM) in order to be executed as it is optimized for longer RNA-seq reads. It is important to keep in mind that STAR is more complex than the other aligners we have seen so far.
Like the previous aligner, STAR first requires an indexed genome:
STAR --runThreadN 8 --runMode genomeGenerate --genomeDir /path/to/genomeDir --genomeFastaFiles genome.fasta --sjdbGTFfile annotations.gtf --sjdbOverhang 100
Notice how it's options are more complicated (for the better!):
-
-runThreadN 8: Number of threads to use. -
-runMode genomeGenerate: Tells STAR to generate a genome index. -
-genomeDir /path/to/genomeDir: Directory to store the index files. -
-genomeFastaFiles genome.fasta: Path to the reference genome FASTA file. -
-sjdbGTFfile annotations.gtf: Path to the gene annotation file (GTF/GFF). -
-sjdbOverhang 100: Read length minus 1 (typically 100 for 101 bp reads).- Note: the author himself, Alexander Dobin has mentioned that this is a horrible name for the function.
sjdbOverhangstands for "Splice Junction Database Overhang", and it determines the length of the genomic sequence around each splice junction that is used during alignment to improve the accuracy of detecting splicing events. This allows STAR to more accurately map reads thatr overlap in exon-exon juctions (in a reference genome, introns are still present, creating potetial mismatches between mRNA and the reference). Difficult concept, but extremely useful in accurate alignments!!!
- Note: the author himself, Alexander Dobin has mentioned that this is a horrible name for the function.
To align single end reads do:
STAR --runThreadN 8 --genomeDir /path/to/genomeDir --readFilesIn reads.fastq --outFileNamePrefix sample_ --outSAMtype BAM SortedByCoordinate
whilst for pair end reads the command is:
STAR --runThreadN 8 --genomeDir /path/to/genomeDir --readFilesIn reads_1.fastq reads_2.fastq --outFileNamePrefix sample_ --outSAMtype BAM SortedByCoordinate
Simiar to Bowtie2, you use a comma to concatenate multiple pair-end reads:
STAR --genomeDir genome_directory --readFilesIn sample1_R1.fastq,sample2_R1.fastq sample1_R2.fastq,sample2_R2.fastq --outFileNamePrefix output_prefix
STAR can also quantify gene expressions using the option --quantMode GeneCounts
STAR --runThreadN 8 --genomeDir /path/to/genomeDir --readFilesIn reads_1.fastq reads_2.fastq --outFileNamePrefix sample_ --outSAMtype BAM SortedByCoordinate --quantMode GeneCounts

Talking about the complications of spliced junctions, HISAT2 is a tool optimized to deal with extactly that. It uses the similar algorithms to the BWA aligner, paired with FM-index, a compression algorithm for improved memory efficiency (unlike STAR!). HISAT2 can handle larger genomes and more complex transcriptomes.
HISAT2 requires an indexed genome, obtainable with a simple synthax:
hisat2-build reference_genome.fa genome_index
Basic synthax:
hisat2 -x genome_index -1 read1.fastq -2 read2.fastq -S output.sam
The options used here are:
-
-x: Specifies the index of the reference genome. -
-1: Specifies the file containing the first reads of paired-end data. -
-2: Specifies the file containing the second reads of paired-end data. -
-S: Specifies the output file in SAM format.
Some other popular options are:
-
-U <single_end_read.fq>: for single end reads -
--dta: Use this option for transcriptome alignments to improve alignments for transcript quantification. -
--rna-strandness: Specify RNA strandness (useFRfor forward,RFfor reverse, orNONEfor no specific strandness). -
--spliced-align: Ensure the aligner considers splicing events. -
--max-intron-len <preferred intron length>: Set the maximum allowed intron length for spliced alignments.
Note
Have you noticed how every single aligner so far outputs files in SAM format?
SAMtools is a suite of tools used for manipulating and analyzing files in the SAM (Sequence Alignment/Map) and BAM (Binary Alignment/Map) formats. These formats are commonly used to store alignment data from various sequencing technologies. SAMtools provides a variety of functionalities to process alignment files, including sorting, indexing, and variant calling.
As mentioned before, SAM and BAM files are essentially the same, with one (SAM, Sequence Alignment/Map, larger size) being human readable whilst the other (BAM, Binary Alignment/Map, more compact) is machine readable.
It is typical to first convert SAM files to BAM as these save space:
samtools view -bS input.sam > output.bam
SAMtools is able to sort SAM/BAM files by the genomic coordinates of the alignments, which is necessary for many downstream analyses:
samtools sort output.bam -o sorted.bam
Typically this is followed by indexing, which creates an index file for BAM files to allow fast access to specific regions of the genome:
samtools index sorted.bam
Users can then create aligment statistics. These can be added to the MultiQC report:
samtools flagstat sorted.bam
At this point, a pileup file can be created for variant calling:
samtools mpileup -f reference.fasta sorted.bam > variants.bcf
A pileup file is a data format used to represent the sequence coverage at each position in the genome across multiple reads. It provides a compact summary of read alignments and is particularly useful for variant calling and other genomic analyses.
It is useful for the following reasons:
- Coverage Information: shows how many reads cover each position in the genome, helping to identify regions with high or low sequencing coverage.
- Can be used for Base Calls: lists the base calls (nucleotides) observed at each position along with their quality scores.
- Variations: allows for the identification of variations such as SNPs (single nucleotide polymorphisms) and indels (insertions and deletions) by comparing observed bases to the reference genome.
Note
Variant calling will be covered in the coming workshop session.
At this point, users have a well aligned sequence in BAM format that can be used for transcriptomic and differential expression analysis. Here are 3 tools that are worth noting (DESeq2 will have its own section later on in the series!).
StringTie is a tool for transcript assembly and quantification from RNA-seq data. It reconstructs transcript structures and estimates their expression levels.
Key Features:
- Transcript Assembly: Assembles transcript models from aligned RNA-seq reads.
- Expression Quantification: Estimates transcript abundance in terms of FPKM (Fragments Per Kilobase of transcript per Million mapped reads) or TPM (Transcripts Per Million).
- Output: Provides GTF/GFF files with transcript annotations and expression levels.
Basic Workflow:
- Input: Aligned reads (BAM files).
- Assembly: Reconstructs transcript models from alignments.
- Quantification: Estimates transcript abundance.
- Output: GTF files with transcript annotations and expression levels.
A simple command to run stringTie is
stringtie aligned_reads.bam -o transcripts.gtf -p 8 -B
where -p is the amount of processors and -B outputs a folder that can be read in Ballgown
Ballgown is an R package used for analyzing and visualizing transcript-level expression data. It works well with data processed by StringTie or other transcript assemblers.
Key Features:
- Differential Expression Analysis: Allows for the comparison of transcript expression between conditions.
- Visualization: Provides tools for visualizing expression data, such as heatmaps and boxplots.
- Integration: Works with GTF files from transcript assemblers and quantification results.
Basic Workflow:
- Input: Transcript-level expression data (e.g., from StringTie) and sample information.
- Data Loading: Imports data into R for analysis.
- Differential Expression: Analyzes differences in transcript expression between conditions.
- Visualization: Creates plots to visualize expression patterns and results.
DESeq2 is a statistical tool used for analyzing count data from RNA-seq experiments to identify differentially expressed genes.
Key Features:
- Differential Expression Analysis: Identifies genes that are significantly differentially expressed between conditions or treatments.
- Normalization: Accounts for differences in sequencing depth and library size by normalizing count data.
- Statistical Modeling: Uses a negative binomial distribution to model count data and perform hypothesis testing. Results Visualization: Provides methods for generating plots such as MA plots and volcano plots to visualize results.
Basic Workflow:
- Input: Raw count data (e.g., from HTSeq or featureCounts) and sample information.
- Normalization: Normalizes data to account for library size.
- Statistical Testing: Performs differential expression analysis using a negative binomial model.
- Output: Results include log fold changes, p-values, and adjusted p-values (e.g., using the Benjamini-Hochberg procedure).