spatialLIBD #1389
Comments
|
Hi @lcolladotor Thanks for submitting your package. We are taking a quick The DESCRIPTION file for this package is: Add SSH keys to your GitHub account. SSH keys |
|
Hi! This is the recently promised package that I mentioned at BioC's slack last Friday.
Having said all that,
Overall, I think this goes beyond a typical As always, if you have any questions, please let me know. I also wanted to let you know that since we are submitting our pre-print soon, likely we'll link to http://research.libd.org/spatialLIBD/ instead of http://bioconductor.org/packages/spatialLIBD unless you have other suggestions. Best, R CMD build/check on R 4.0.0
BiocCheck on R 3.6.2Below is the output of $ R CMD BiocCheck --new-package spatialLIBD_0.99.0.tar.gz
This is BiocCheck version 1.22.0. BiocCheck is a work in progress.
Output and severity of issues may change. Installing package...
* Checking Package Dependencies...
* Checking if other packages can import this one...
* Checking to see if we understand object initialization...
* Checking for deprecated package usage...
* Checking for remote package usage...
* Checking version number...
* Checking for version number mismatch...
* Checking new package version number...
* Checking R Version dependency...
* Checking package size...
* Checking individual file sizes...
* Checking biocViews...
* Checking that biocViews are present...
* Checking package type based on biocViews...
ExperimentData
* Checking for non-trivial biocViews...
* Checking that biocViews come from the same category...
* Checking biocViews validity...
* Checking for recommended biocViews...
* NOTE: Consider adding these automatically suggested biocViews:
PackageTypeData
See http://bioconductor.org/developers/how-to/biocViews/
* Checking build system compatibility...
* Checking for blank lines in DESCRIPTION...
* Checking if DESCRIPTION is well formatted...
* Checking for whitespace in DESCRIPTION field names...
* Checking that Package field matches directory/tarball name...
* Checking for Version field...
* Checking for valid maintainer...
* Checking DESCRIPTION/NAMESPACE consistency...
This is not a software package, skipping vignette checks...
* Checking library calls...
* Checking for library/require of spatialLIBD...
* Checking coding practice...
* NOTE: Avoid sapply(); use vapply()
Found in files:
check_modeling_results.R (line 33, column 9)
check_modeling_results.R (line 40, column 19)
check_modeling_results.R (line 41, column 13)
gene_set_enrichment.R (line 80, column 17)
gene_set_enrichment.R (line 100, column 26)
gene_set_enrichment.R (line 101, column 28)
layer_boxplot.R (line 192, column 17)
layer_stat_cor.R (line 56, column 17)
sig_genes_extract.R (line 78, column 29)
sig_genes_extract.R (line 93, column 9)
sig_genes_extract.R (line 97, column 9)
sig_genes_extract.R (line 101, column 9)
* NOTE: Avoid system() ; use system2()
Found in files:
inst/scripts/make-data_spatialLIBD.R (line 201)
inst/scripts/make-data_spatialLIBD.R (line 218)
inst/doc/spatialLIBD.Rmd (line 192)
vignettes/spatialLIBD.Rmd (line 192)
* WARNING: Remove set.seed usage in R code
Found in R/ directory functions:
layer_boxplot()
* Checking parsed R code in R directory, examples, vignettes...
* Checking function lengths...................................................
* NOTE: Recommended function length <= 50 lines.
There are 14 functions > 50 lines.
The longest 5 functions are:
app_server() (R/app_server.R, line 17): 1273 lines
app_ui() (R/app_ui.R, line 6): 516 lines
layer_boxplot() (R/layer_boxplot.R, line 104): 123 lines
check_sce() (R/check_sce.R, line 25): 89 lines
layer_matrix_plot() (R/layer_matrix_plot.R, line 59): 86 lines
* Checking man page documentation...
* Checking package NEWS...
* Checking unit tests...
* Checking skip_on_bioc() in tests...
* Checking formatting of DESCRIPTION, NAMESPACE, man pages, R source,
and vignette source...
* NOTE: Consider shorter lines; 305 lines (5%) are > 80 characters
long.
First 6 lines:
R/app_server.R:37 ## From /dcl02/lieber/ajaffe/SpatialTranscriptomi...
R/app_server.R:38 # cat(paste0("'", names(cols_layers_martinowich),...
R/app_server.R:74 } else if (input$cluster %in% c('layer_guess'...
R/app_server.R:76 } else if (input$cluster %in% c('layer_guess_...
R/app_server.R:356 assays(sce_sub)[[assayname]][which(r...
R/app_server.R:413 scale_fill_manual(values = get_colors(co...
* NOTE: Consider multiples of 4 spaces for line indents, 118
lines(2%) are not.
First 6 lines:
man/check_sce.Rd:8 sce,
man/check_sce.Rd:9 variables = c("GraphBased", "Layer", "Maynard", "M...
man/fetch_data.Rd:8 type = c("sce", "sce_layer", "modeling_results"),
man/fetch_data.Rd:9 destdir = tempdir(),
man/fetch_data.Rd:10 eh = ExperimentHub::ExperimentHub()
man/gene_set_enrichment_plot.Rd:8 enrichment,
See http://bioconductor.org/developers/how-to/coding-style/
See FormatR package:
https://cran.r-project.org/web/packages/formatR/index.html
* Checking if package already exists in CRAN...
* Checking if new package already exists in Bioconductor...
* Checking for bioc-devel mailing list subscription...
* NOTE: Cannot determine whether maintainer is subscribed to the
bioc-devel mailing list (requires admin credentials). Subscribe
here: https://stat.ethz.ch/mailman/listinfo/bioc-devel
* Checking for support site registration...
Maintainer is registered at support site.
Summary:
ERROR count: 0
WARNING count: 1
NOTE count: 7
For detailed information about these checks, see the BiocCheck
vignette, available at
https://bioconductor.org/packages/3.10/bioc/vignettes/BiocCheck/inst/doc/BiocCheck.html#interpreting-bioccheck-output |
|
A reviewer has been assigned to your package. Learn what to expect IMPORTANT: Please read the instructions for setting |
|
Dear Package contributor, This is the automated single package builder at bioconductor.org. Your package has been built on Linux, Mac, and Windows. On one or more platforms, the build results were: "skipped, ERROR". Please see the build report for more details. |
|
Received a valid push; starting a build. Commits are: 7ac24dc v0.99.10 -- update AWS file locations, resolve a b... |
|
Dear Package contributor, This is the automated single package builder at bioconductor.org. Your package has been built on Linux, Mac, and Windows. On one or more platforms, the build results were: "ERROR". Please see the build report for more details. |
|
Hi @dvantwisk. Thank you for reviewing If that's not the case, please let me know. This is related to eddelbuettel/rcppannoy#57. In any case, it looks from eddelbuettel/rcppannoy#57 (comment) that As for the BiocCheck warnigns about R 3.6 and Best, |
|
For |
|
Received a valid push; starting a build. Commits are: 5024450 v0.99.11 -- test without listing the version of Rc... |
|
Dear Package contributor, This is the automated single package builder at bioconductor.org. Your package has been built on Linux, Mac, and Windows. On one or more platforms, the build results were: "ERROR". Please see the build report for more details. |
|
Received a valid push; starting a build. Commits are: a2c0d08 v0.99.12 -- see NEWS.md for details |
|
Dear Package contributor, This is the automated single package builder at bioconductor.org. Your package has been built on Linux, Mac, and Windows. On one or more platforms, the build results were: "ERROR, WARNINGS". Please see the build report for more details. |
|
Hi, So I moved I did reduce the number of NOTEs from 6 to 3 for BiocCheck. The error by As for Windows, R CMD check fails due to memory. How much memory does that builder have? Because the package needs about 2.5 GB of RAM to load the data it provides. Best, |
|
but generally reproducibility involving random numbers depends on the order in which the user invokes functions, how is this any different? |
|
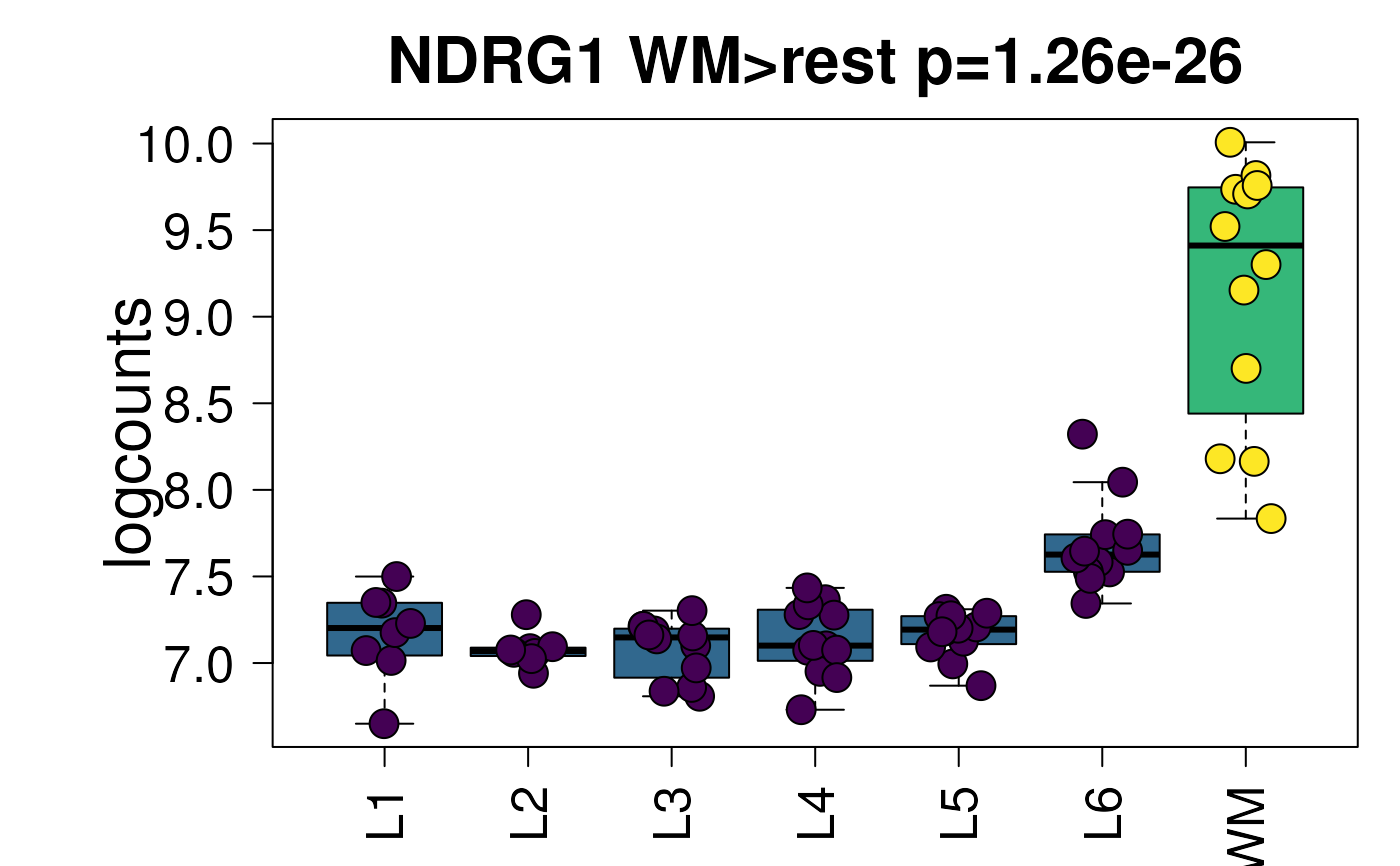
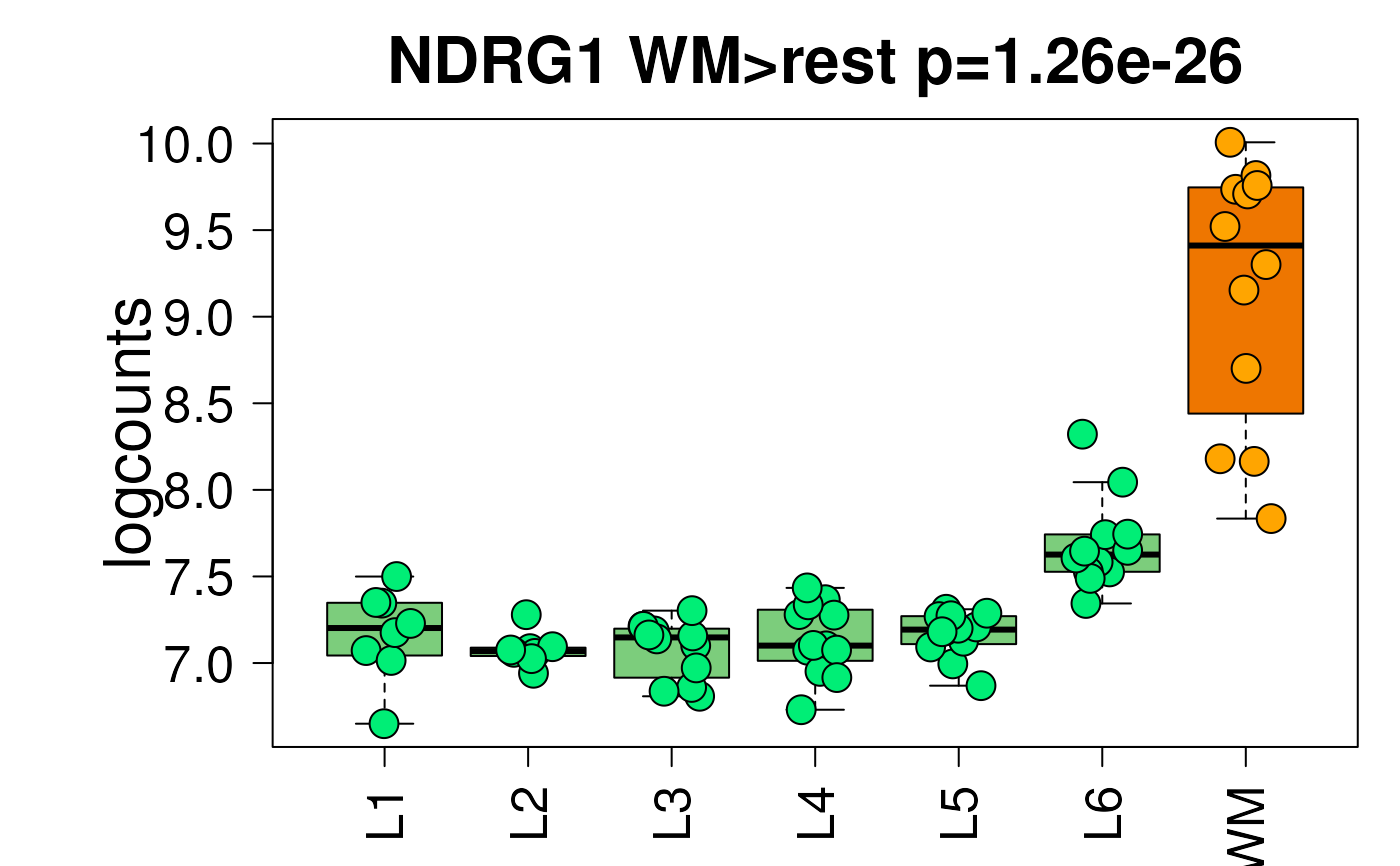
I simply want the boxplots to have points plotted on top of them and to be jittered randomly, but controlled in such a way that you can re-make the same plot that you see in the shiny app through the R console. That way you can make it on the R console and save to a different set of pdf size/height if you want to. Or you can make the sample plot and change the colors as in http://research.libd.org/spatialLIBD/reference/layer_boxplot-5.png and http://research.libd.org/spatialLIBD/reference/layer_boxplot-6.png. Originally, I was able to do this by having a If a user (or myself) runs |
{kind=link}
{kind=link}
|
The best advice is to not call set.seed in your package code. Continue to do so if you insist, but know that this deviates from best practice. |
|
I apologize for the silence on my end. Even though we are still deliberating about some of the errors you are having on our builders' GENERAL
vignettes/spatialLIBD.R
|
|
Regarding the issues you've been having with the builder:
|
|
ohh, it's been 6 days =( Sorry, I was teaching a course last week and I'm behind on several projects. Thank you for looking into the package! I'll work on making a smaller SCE object at the spot-level (the 2+ GB one currently) just for some plots and use that for the examples in order to get through the 2.5 GB limit on the Windows builder (I didn't know about that limit, thanks for the info!). I already have one but it was kind of used internally only in the vignette (and not in the R help files) which is why there are some non-shiny chunks with Best, |
|
Hello, I just want to check in with the progress of your package submission. I need to send you a reminder that the deadline for your package to be included in the next Bioconductor release is April 21st. The package will need to be accepted by then to be included in the next release. Please message me back if you have any issues making the changes I specified. |
|
I do need to remind you that the final day to accept packages for the upcoming Bioconductor release is April 22nd. Please message me if you have any other questions. |
|
Received a valid push; starting a build. Commits are: 8970f02 v0.99.13 -- Switch to GitHub actions as described ... |
|
Dear Package contributor, This is the automated single package builder at bioconductor.org. Your package has been built on Linux, Mac, and Windows. On one or more platforms, the build results were: "WARNINGS, ERROR". Please see the build report for more details. |
|
Hi, Sorry for dropping the ball on the communicating here. I got really into GitHub Actions at r-lib/actions#84 since Wednesday last week so I got behind on this project. Anyway, I'm working on This is in relation to the github repo size that you mentioned in #1389 (comment). Steps that I followed
$ git clone [email protected]:LieberInstitute/spatialLIBD.git
Cloning into 'spatialLIBD'...
remote: Enumerating objects: 742, done.
remote: Counting objects: 100% (742/742), done.
remote: Compressing objects: 100% (365/365), done.
remote: Total 1621 (delta 364), reused 675 (delta 325), pack-reused 879
Receiving objects: 100% (1621/1621), 33.50 MiB | 6.28 MiB/s, done.
Resolving deltas: 100% (907/907), done.
$ cd spatialLIBD/
$ du -sh .
44M .
Bioconductor as a wholeHence, I think that this could affect Bioconductor at large if the SBP and the daily builds just clone the branch needed (so git clone -b master --single-branch [email protected]:LieberInstitute/spatialLIBD.gitBack to
|
|
Dear Package contributor, This is the automated single package builder at bioconductor.org. Your package has been built on Linux, Mac, and Windows. On one or more platforms, the build results were: "WARNINGS". Please see the build report for more details. |
|
Received a valid push; starting a build. Commits are: 05664de v0.99.15 -- fix a NOTE |
|
Hi, I'm happy to report that the package There are still two
This version ( Version Best, * Using R 4.1 and not 4.0 on both macOS and Windows due to the weird nature of this window of time where there's more versions of R beyond |
|
Dear Package contributor, This is the automated single package builder at bioconductor.org. Your package has been built on Linux, Mac, and Windows. On one or more platforms, the build results were: "WARNINGS". Please see the build report for more details. |
|
Your package has been accepted. It will be added to the Thank you for contributing to Bioconductor! |
|
The master branch of your GitHub repository has been added to Bioconductor's git repository. To use the git.bioconductor.org repository, we need an 'ssh' key to associate with your github user name. If your GitHub account already has ssh public keys (https://github.com/lcolladotor.keys is not empty), then no further steps are required. Otherwise, do the following: See further instructions at https://bioconductor.org/developers/how-to/git/ for working with this repository. See especially https://bioconductor.org/developers/how-to/git/new-package-workflow/ to keep your GitHub and Bioconductor repositories in sync. Your package will be included in the next nigthly 'devel' build (check-out from git at about 6 pm Eastern; build completion around 2pm Eastern the next day) at https://bioconductor.org/checkResults/ (Builds sometimes fail, so ensure that the date stamps on the main landing page are consistent with the addition of your package). Once the package builds successfully, you package will be available for download in the 'Devel' version of Bioconductor using https://bioconductor.org/packages/spatialLIBD If you have any questions, please contact the bioc-devel mailing list (https://stat.ethz.ch/mailman/listinfo/bioc-devel); this issue will not be monitored further. |
Update the following URL to point to the GitHub repository of
the package you wish to submit to Bioconductor
Confirm the following by editing each check box to '[x]'
I understand that by submitting my package to Bioconductor,
the package source and all review commentary are visible to the
general public.
I have read the Bioconductor Package Submission
instructions. My package is consistent with the Bioconductor
Package Guidelines.
I understand that a minimum requirement for package acceptance
is to pass R CMD check and R CMD BiocCheck with no ERROR or WARNINGS.
Passing these checks does not result in automatic acceptance. The
package will then undergo a formal review and recommendations for
acceptance regarding other Bioconductor standards will be addressed.
My package addresses statistical or bioinformatic issues related
to the analysis and comprehension of high throughput genomic data.
I am committed to the long-term maintenance of my package. This
includes monitoring the support site for issues that users may
have, subscribing to the bioc-devel mailing list to stay aware
of developments in the Bioconductor community, responding promptly
to requests for updates from the Core team in response to changes in
R or underlying software.
I am familiar with the essential aspects of Bioconductor software
management, including:
months, for bug fixes.
(optionally via GitHub).
For help with submitting your package, please subscribe and post questions
to the bioc-devel mailing list.
The text was updated successfully, but these errors were encountered: