4. Step by step introduction
Generally, using the three functions runScStatistics, runScAnnotation and runScCombination introduced in last section can generate detailed graphical HTML reports to make users have a quick overview for the data. If users want to understand the meaning of each argument in all steps, they can read the following introductions.
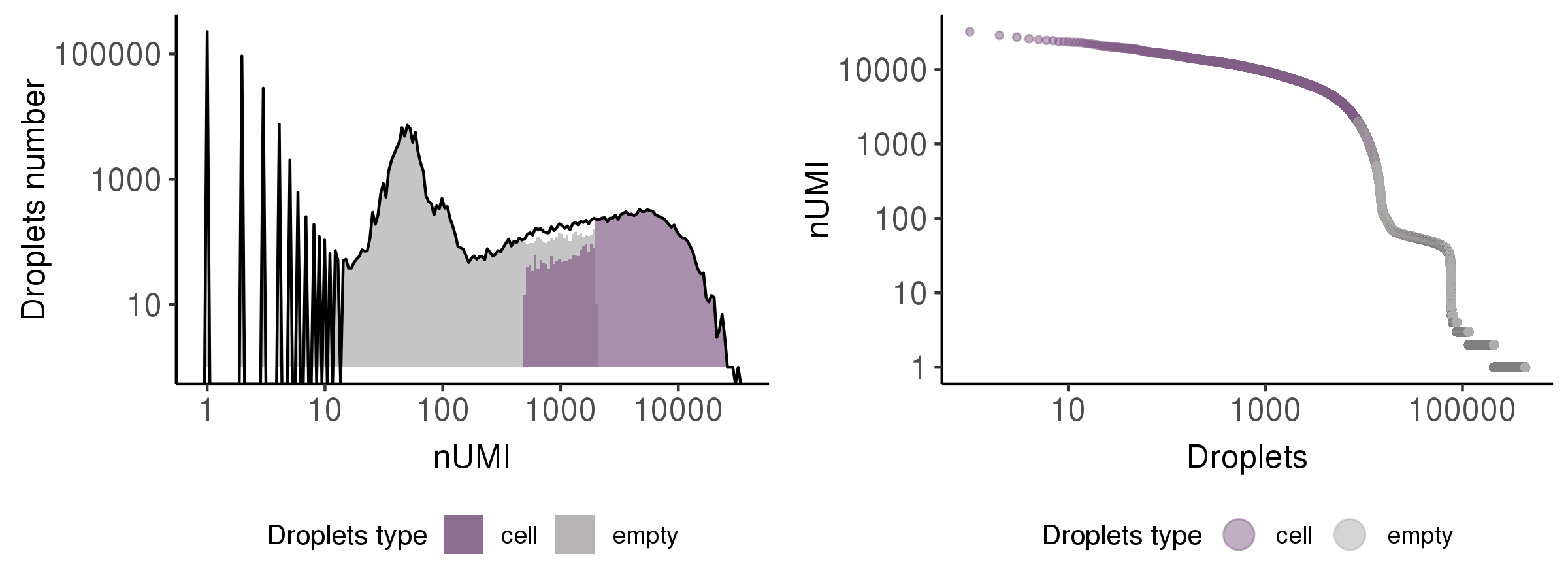
This step is to identify the droplets more likely to include real cells.
Generally, Cell Ranger V3 performs this step and shows good performance.
In this step, we showed the results by a histogram and a rank plot to present the distribution of total UMI counts (nUMI) in putative cells (purple) and empty droplets (grey)s.
Ideally, one droplet contains one cell in good state and the detected RNA transcripts are all from this cell. However, some abnormal situations may occur, so we calculate following metrics to perform cell quality control (QC).
-
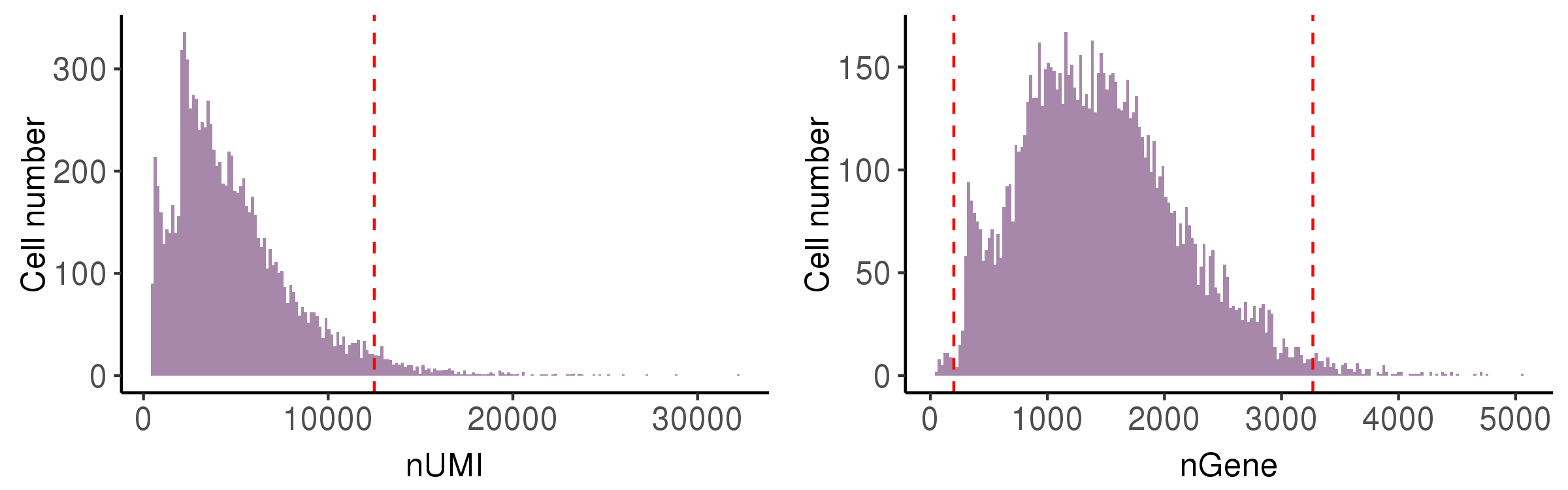
nUMI: the number of total UMIs in the droplet. Too small means no cells are captured, and too large means capturing two or more. -
nGene: the number of expressed genes in the droplet. Too small means the loss of transcripts diversity. Too large means containing two or more cells. -
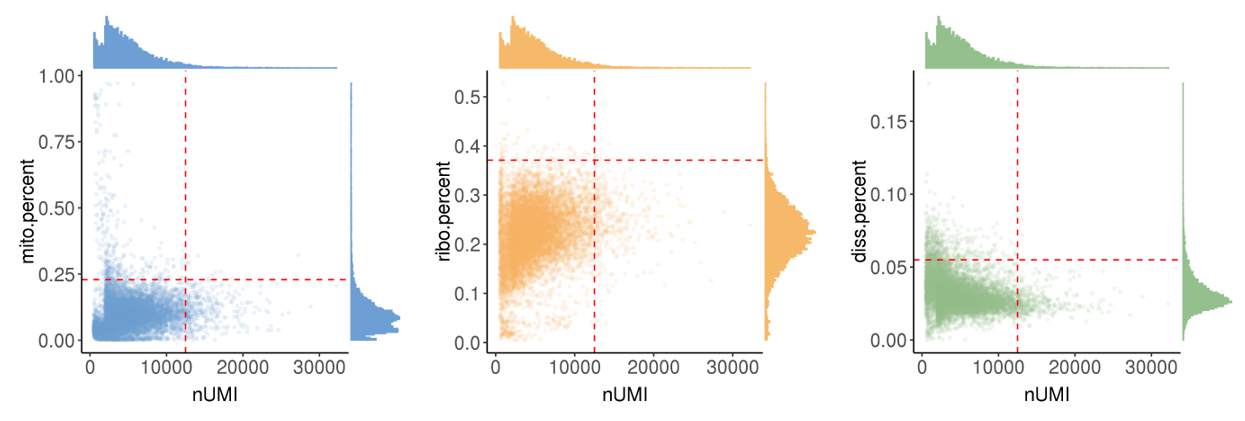
mito.percent: the percentage of UMIs from mitochondrial genes. Too large means the captured cell is necrotic or lysed. -
ribo.percent: the percentage of UMIs from ribosome genes. Too large means the captured cell is necrotic or lysed. -
diss.percent: the percentage of UMIs from dissociation-associated genes. Too large means dissociation process has serious effects on the cell states.
In this step, we showed the distribution of these metrics and provide automatically identified filter thresholds.
The runScAnnotation performs finial cell filter by according to the thresholds recorded in file cell.QC.thres.txt, which is generated by scStatistics. So users can modify the values in the file to adjust the strength of QC. At the same time, the argument bool.filter.cell of function runScAnnotation can control whether to filter cells.
For quality control on genes, we firstly filtered genes which expressed in less than 3 cells. Then, considering that some necrotic or lysed cells may leak their RNA transcripts into the external suspensions, and lead to other droplets being contaminated by these ambient RNA transcripts, we also performed some statistical analyses on the influence of contamination.
In this step, we calculated following three metrics.
-
bg.percent: the expression proportion for each gene in background distribution (all droplets withnUMI <= 10) -
prop.median: the median of expression proportions for a gene in each cell. -
detect.rate: the detected (#UMI > 0) rate for a gene in all cells.
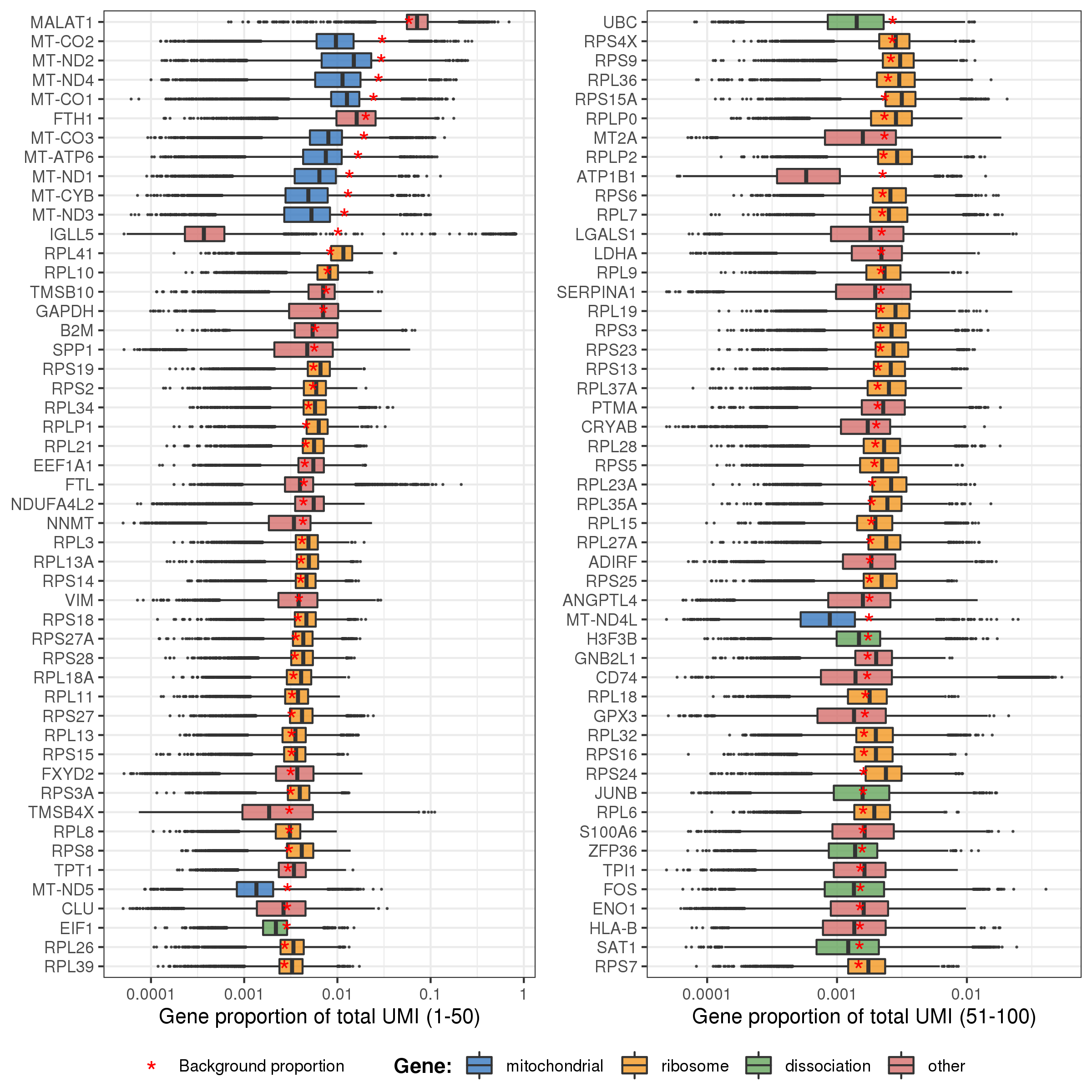
The plot below shows the distributions of gene proportion in cells for the first 100 genes (ordered by their proportion in background bg.percent). And the points (genes) are colored according to whether they belongs to mitochondrial, ribosome, or dissociation associated genes. The red star signs mark the genes’ proportion in background.
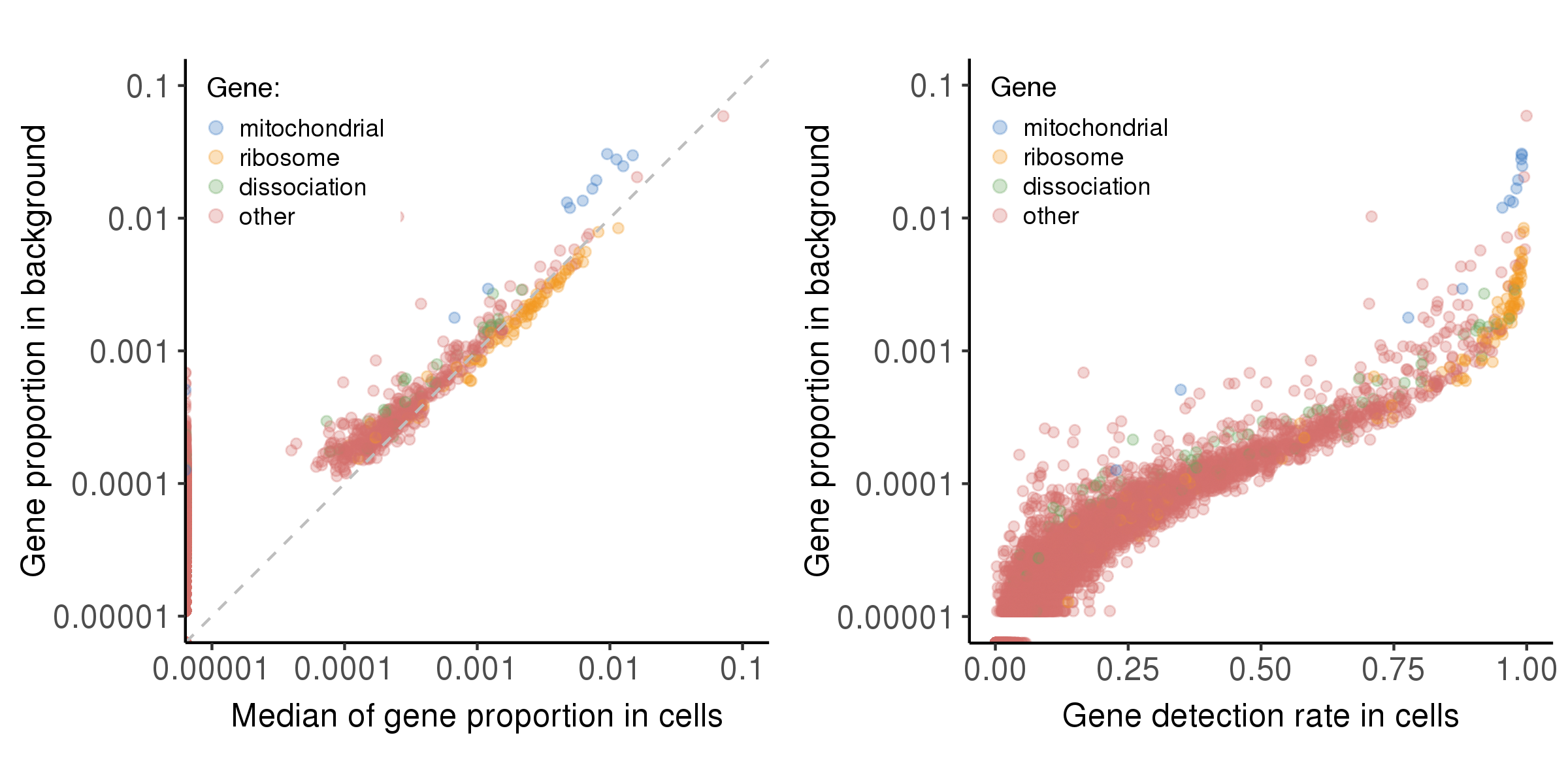
The plot below shows the relationship between bg.percent and prop.median, bg.percent and detect.rate.
The argument bool.filter.gene of function runScAnnotation can control whether to filter genes. If it is TRUE, the argument anno.filter can determine what kind of genes (the default is c("mitochondrial", "ribosome", "dissociation")) are filtered. The argument nCell.min and bgPercent.max can be used to control the gene filter strength of the metrics nCell and bg.percent.
Besides, we also integrated the package SoupX
to estimate the contamination fraction of ambient RNAs from lysed cells.
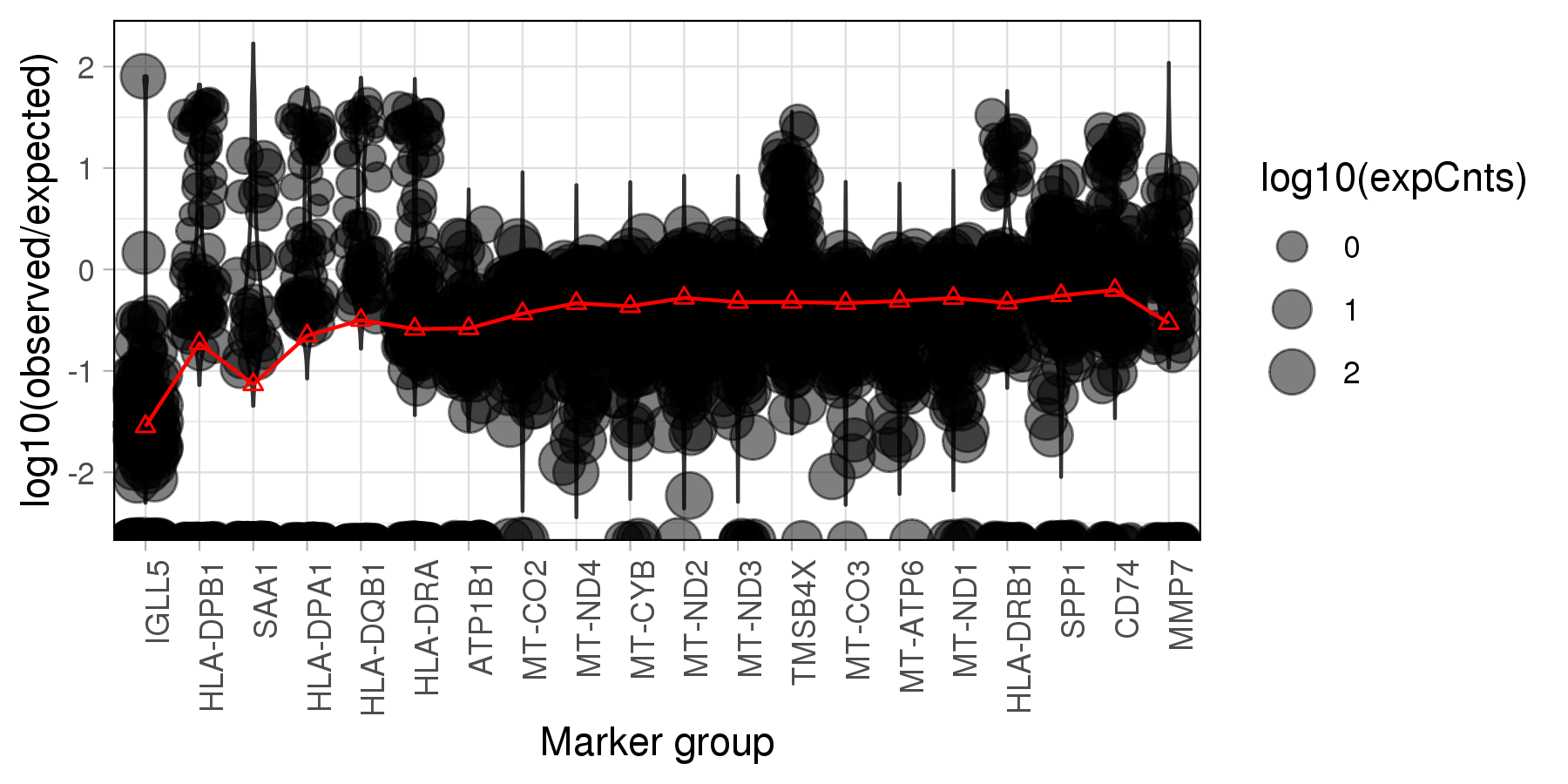
The plot below is generated by SoupX, which visualises the log10 ratios of observed expression counts to expected if the cell is pure background. The read lines marks the estimated contamination fraction using each genes.
Note: The SoupX emphasize that the genes
in the plot are heuristic and are just used to help develop biological intuition.
It absolutely must not be used to automatically select the top N genes from the list,
which may over-estimate the contamination fraction!
By default, we set three default gene sets (immunoglobulin, haemoglobin, and MHC genes)
according to the characteristics of cancer microenvironment. Users can input their seleted genes via argument bg.spec.genes to the function runScStatistics. And the argument bool.runSoupx can control whether to perform this step.
Then in scAnnotation module, users can use bool.rmContamination (default is FALSE) to control whether to remove ambient RNA contamination based on SoupX. If it is TRUE, the argument contamination.fraction determines the estimated contamination fraction. If contamination.fraction is NULL, the result of scStatistics will be used.
The basic analyses of single cell data are mainly performed by Seurat, which included normalization, log-transformation, highly variable genes identification, unwanted variance removing, scaling, centering, dimension reduction (PCA/t-SNE/UMAP), clustering, and differential expression analysis.
In runScAnnotation, following arguments can determine detailed setting of these steps.
-
vars.add.metaindicates the variables to be added to Seurat object'smeta.data.The default isc("mito.percent", "ribo.percent", "diss.percent"). -
vars.to.regressindicates the variables to regress out in Seurat. The default isc("nUMI", "mito.percent", "ribo.percent"). The argumentpc.useindicats the number of PCs to use. The default value is30. -
resolutioncontrols the strength of clustering. The default is 0.8. -
clusterStashNameindicates the recorded name of cluster identies. The default is "default". -
show.featuresindicates the other users interested marker genes to be plotted. -
bool.add.featuresdetermines whether to add default marker genes toshow.features. -
bool.runDiffExprdetermines whether to perform differential expressed analysis. -
n.markersdetermines the number of differential expressed genes showed in the heatmap. The defalut is 5.
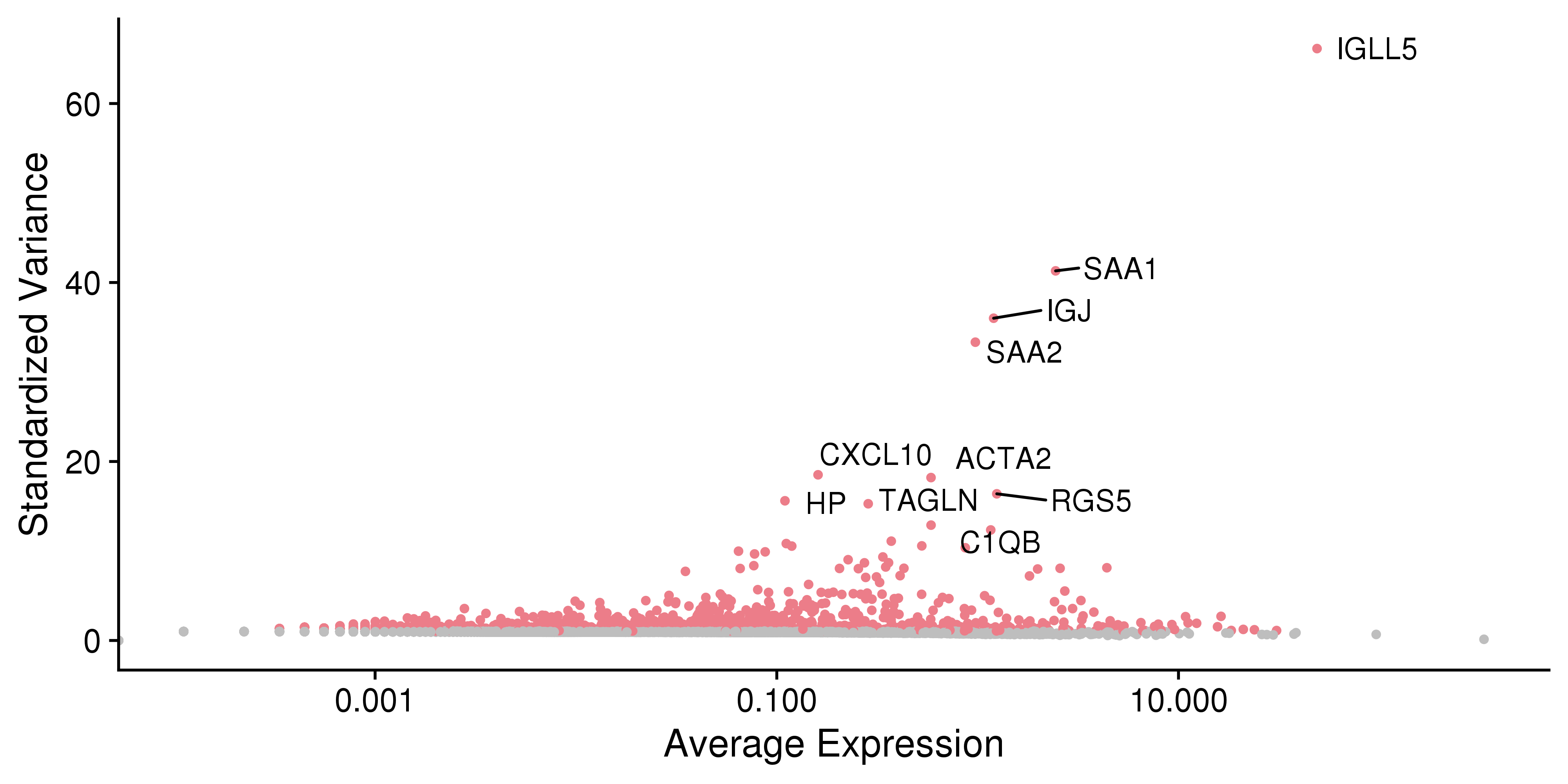
This plot is for highly variable genes.
This plot is for common marker genes.
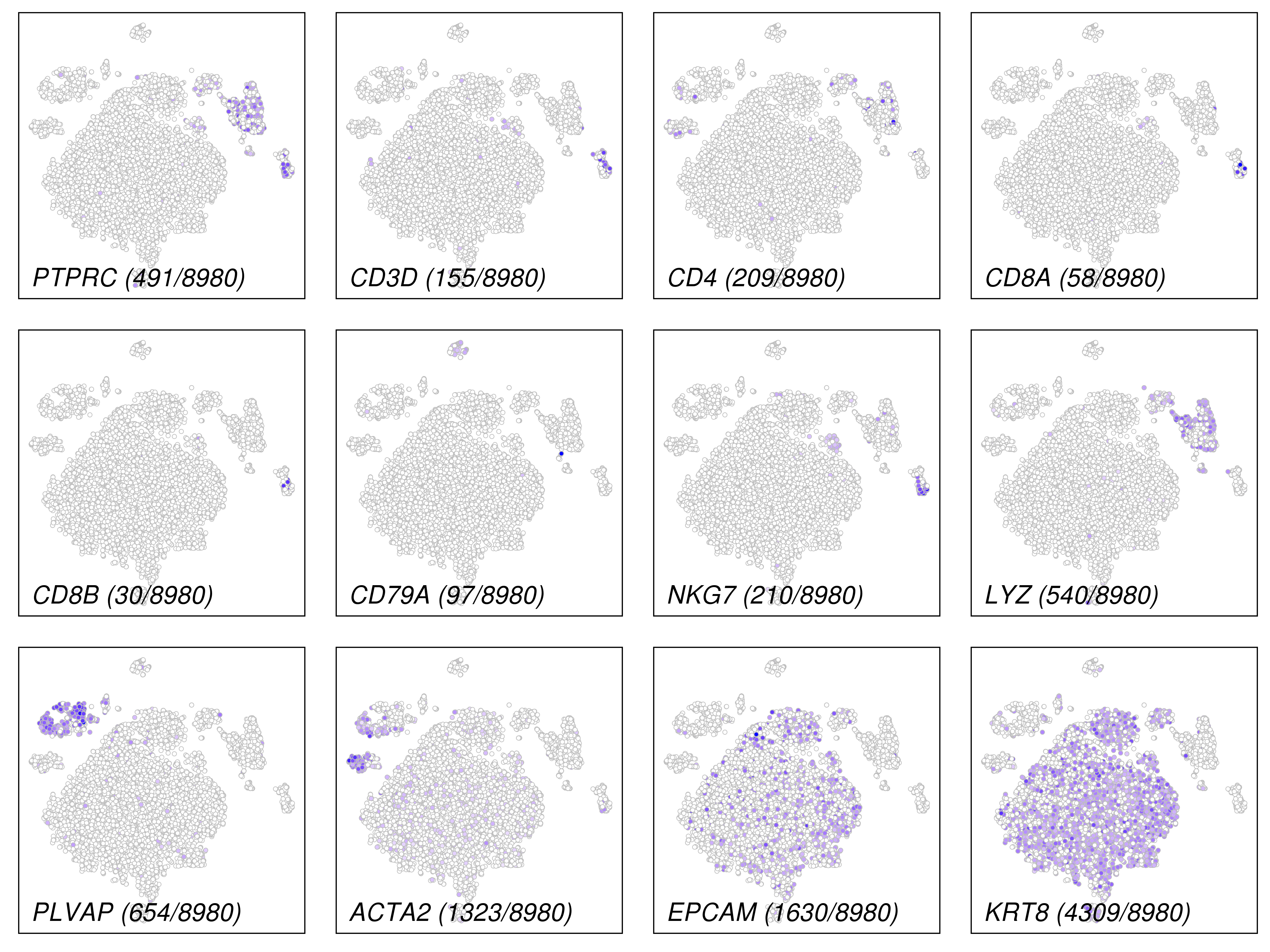
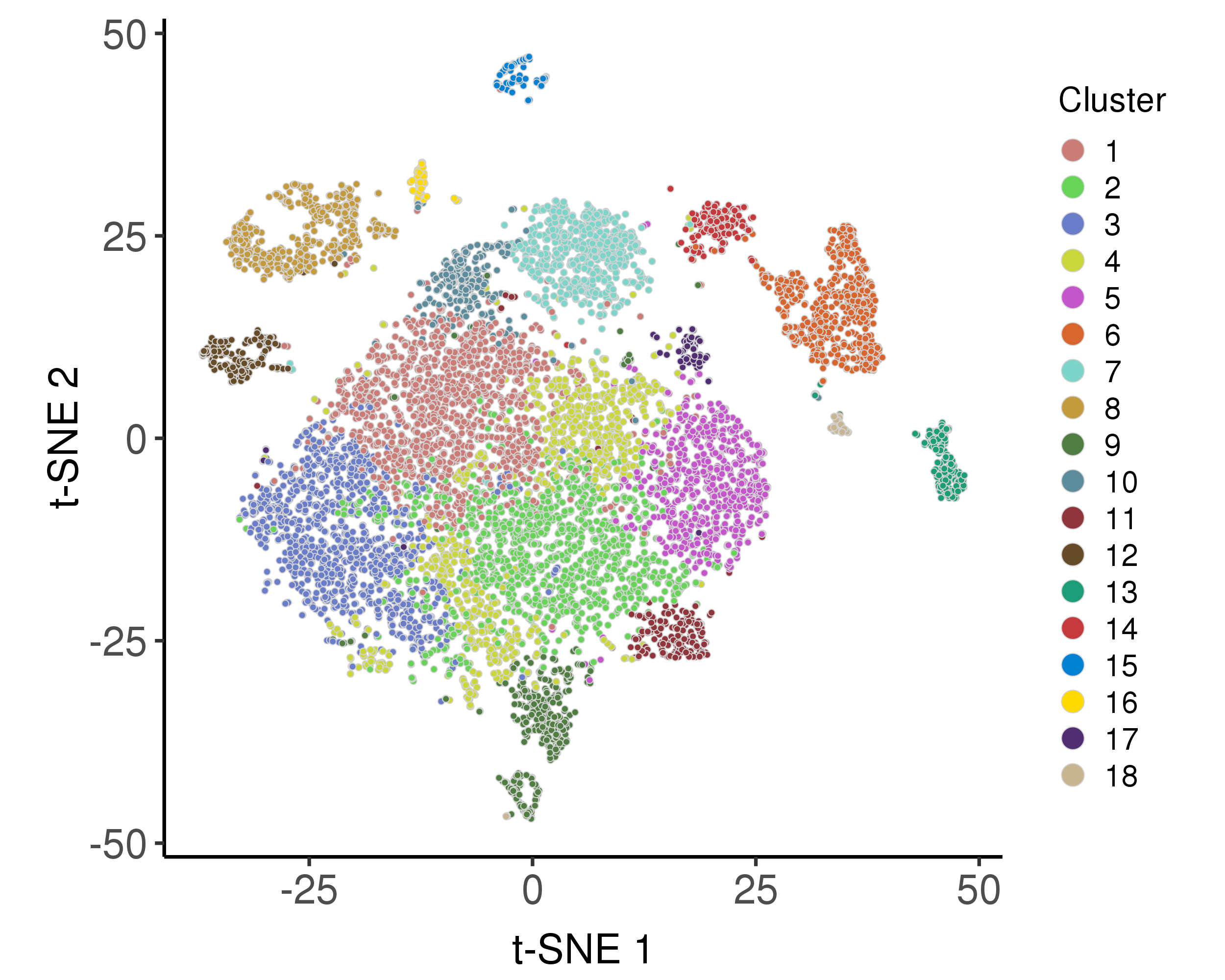
These plots are for clustering on t-SNE and UMAP 2D space.

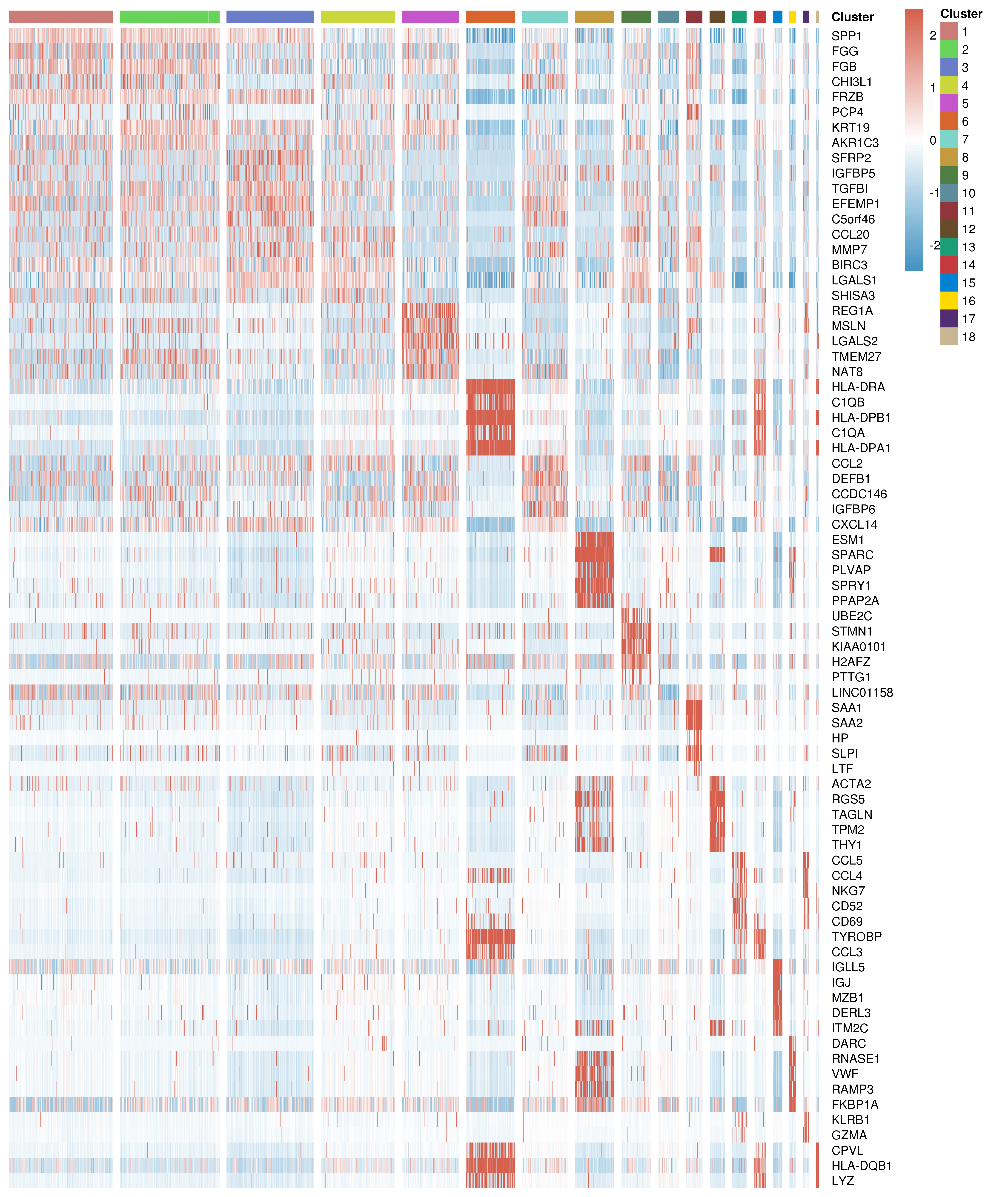
This plot is for differnetial expression analysis.
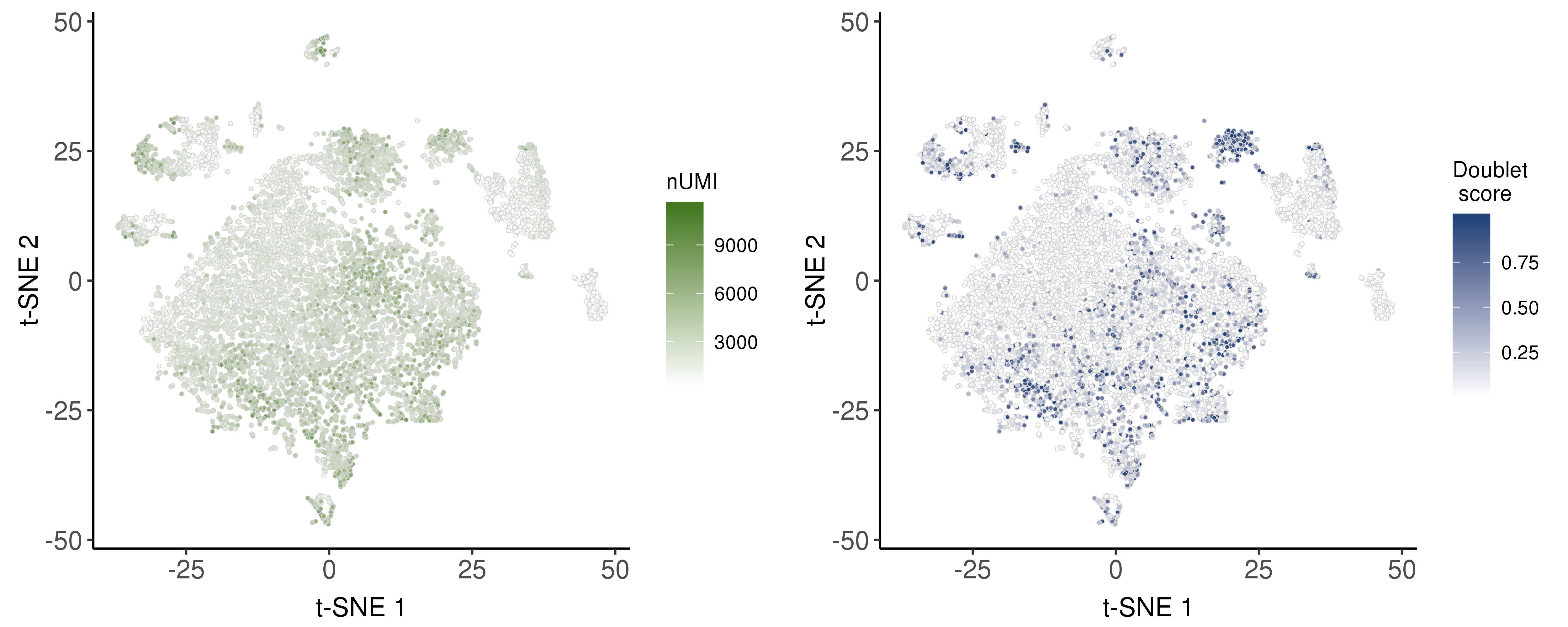
We estimated doublet score based on the package scds.
In runScAnnotation, following arguments can determine detailed setting of this step.
-
bool.runDoubletindicates whether to perform this step. -
doublet.methodindicates the method to estimate doublet score. The default is "cxds". "cxds"(co-expression based doublet scoring) and "bcds"(binary classification based doublet scoring) are allowed.
Following are the distribution of nUMI and doublet scores.
We used one-class logistic regression (OCLR) model to predict common cancer micro-environmental cell types.
In runScAnnotation, following arguments can determine detailed setting of this step.
-
bool.runCellClassifyindicates whether to predict the usual cell type. The default isTRUE. -
ct.templatesindicates OCLR cell type templates used to classification. The default is NULL and eight default templates, including endothelial cells, fibroblasts, and immune cells (CD4+ T cells, CD8+ T cells, B cells, nature killer cells, and myeloid cells) will be used. Users can also train their own templates (the method can be found in nextOther personalized settingssection.
Following are the distribution of the predicted cell types.
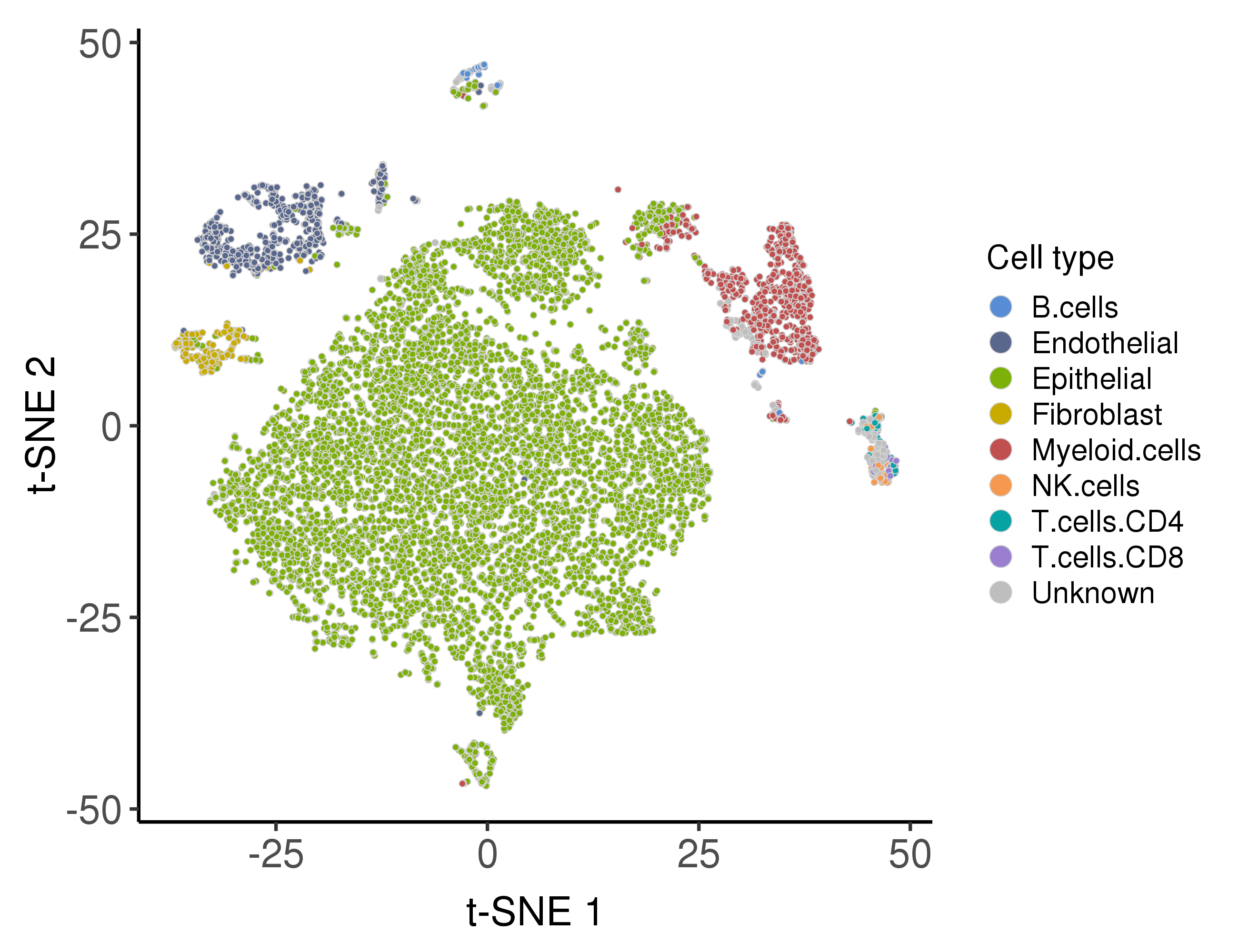
The malignancy scores are based on infercnv algorithm.
In runScAnnotation, following arguments can determine detailed setting of this step.
-
bool.runMalignancyindicates whether to estimate malignancy. -
cnv.ref.dataindicates the expression matrix used as the normal reference. The default isNULL, and an default normal data will be used. User can also input their own reference data by it. -
cnv.referAdjMatindicates the adjacent matrix for the normal reference data. The larger the value, the closer the cell pair is. The default is NULL, and a SNN matrix of the defaultcnv.ref.datawill be used. -
cutoffis a threshold used in the CNV inference.
Following is the distribution of the estimated malignancy scores.
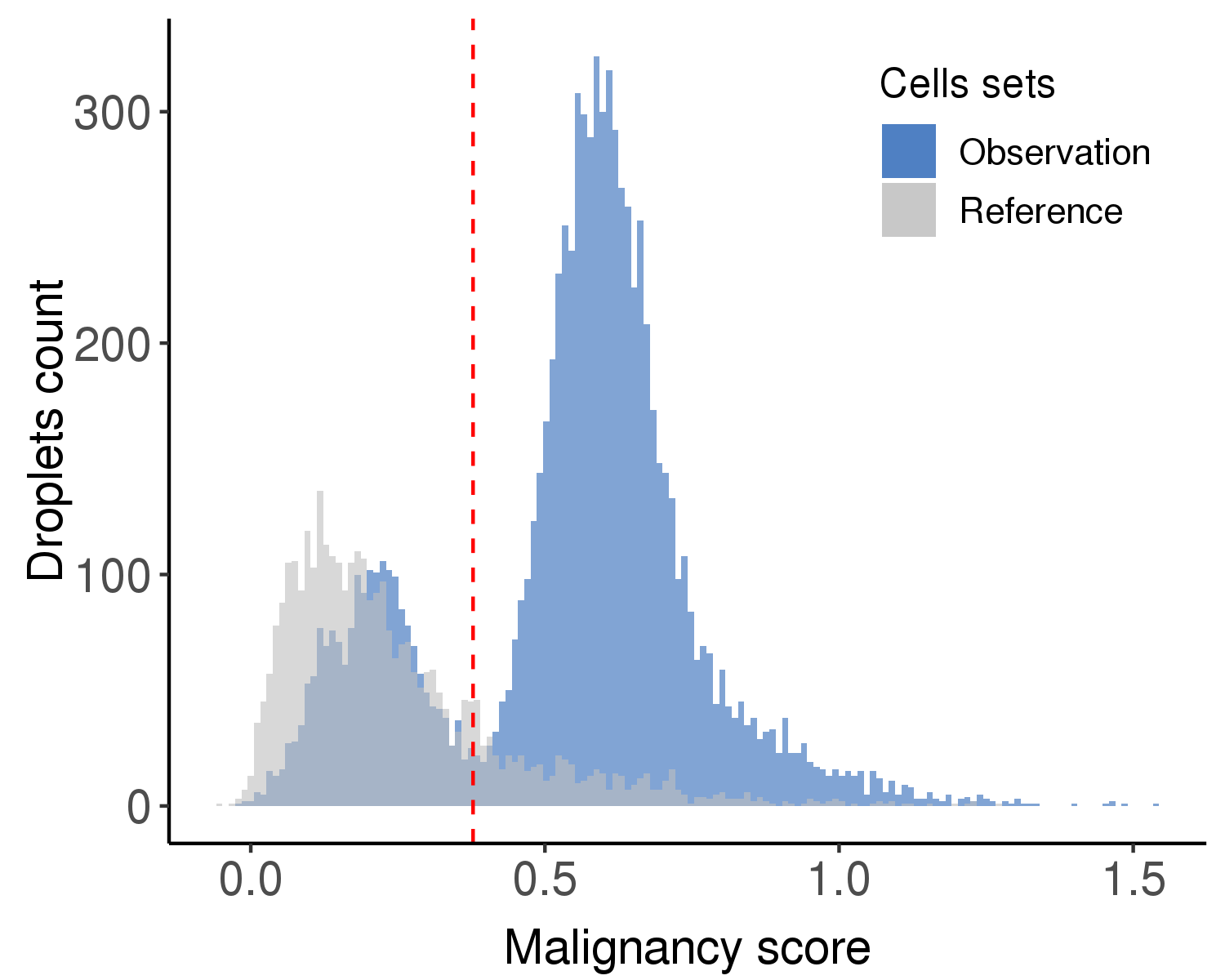
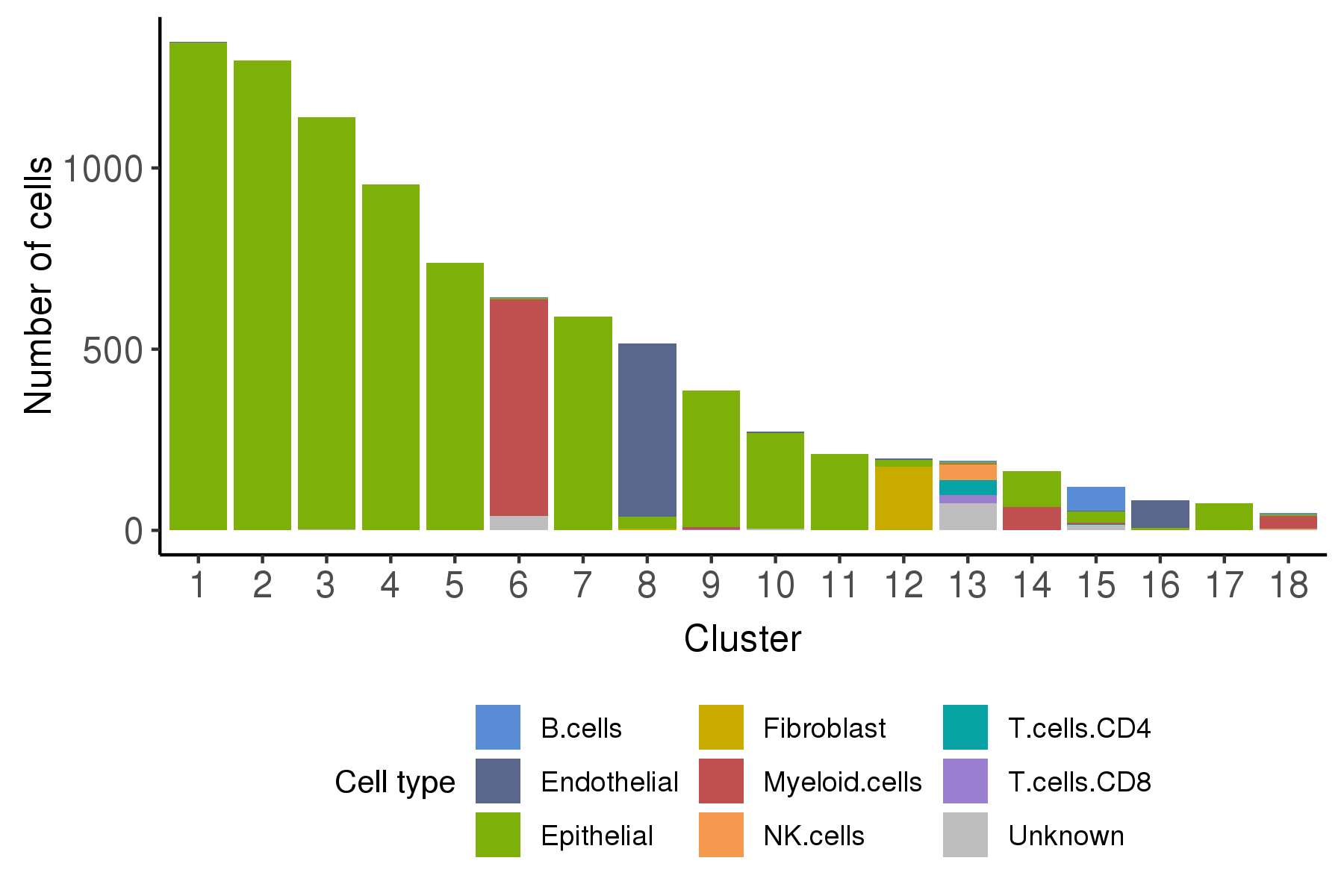
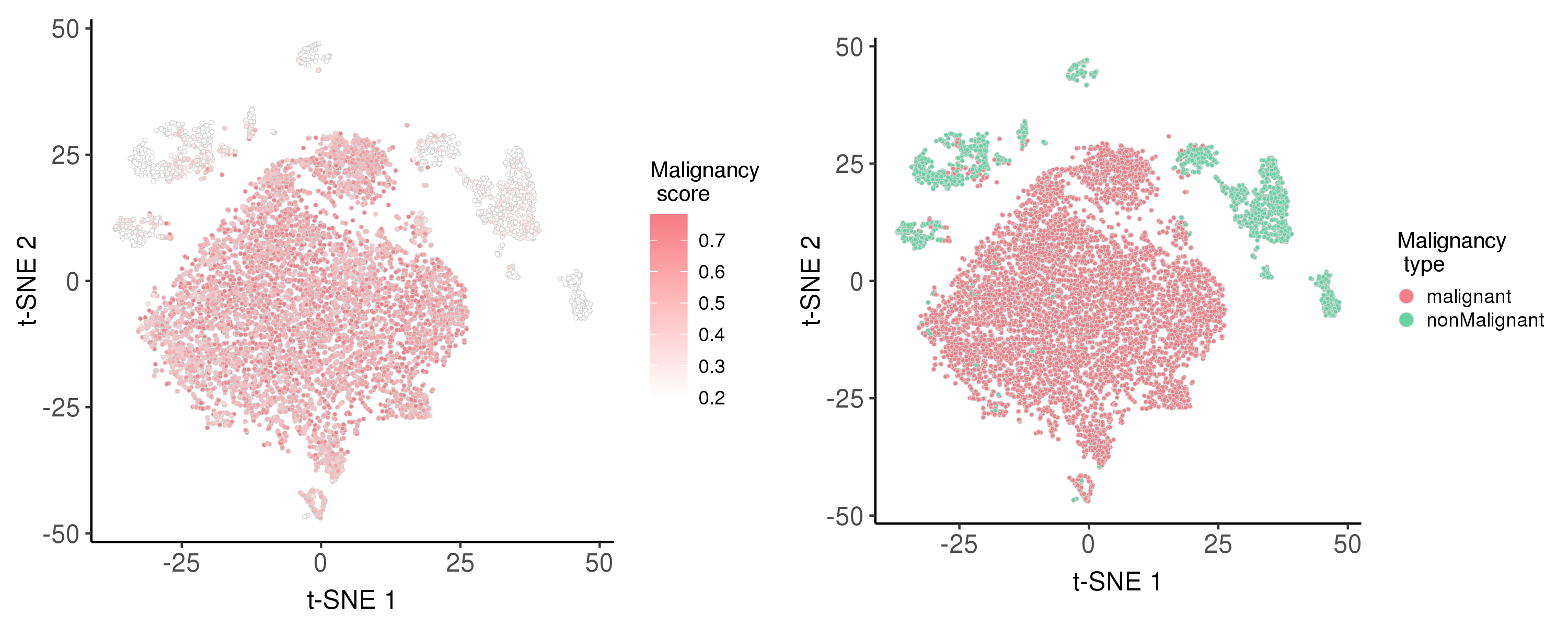
Following is the t-SNE plot colored by malignancy score (left) and type (right).
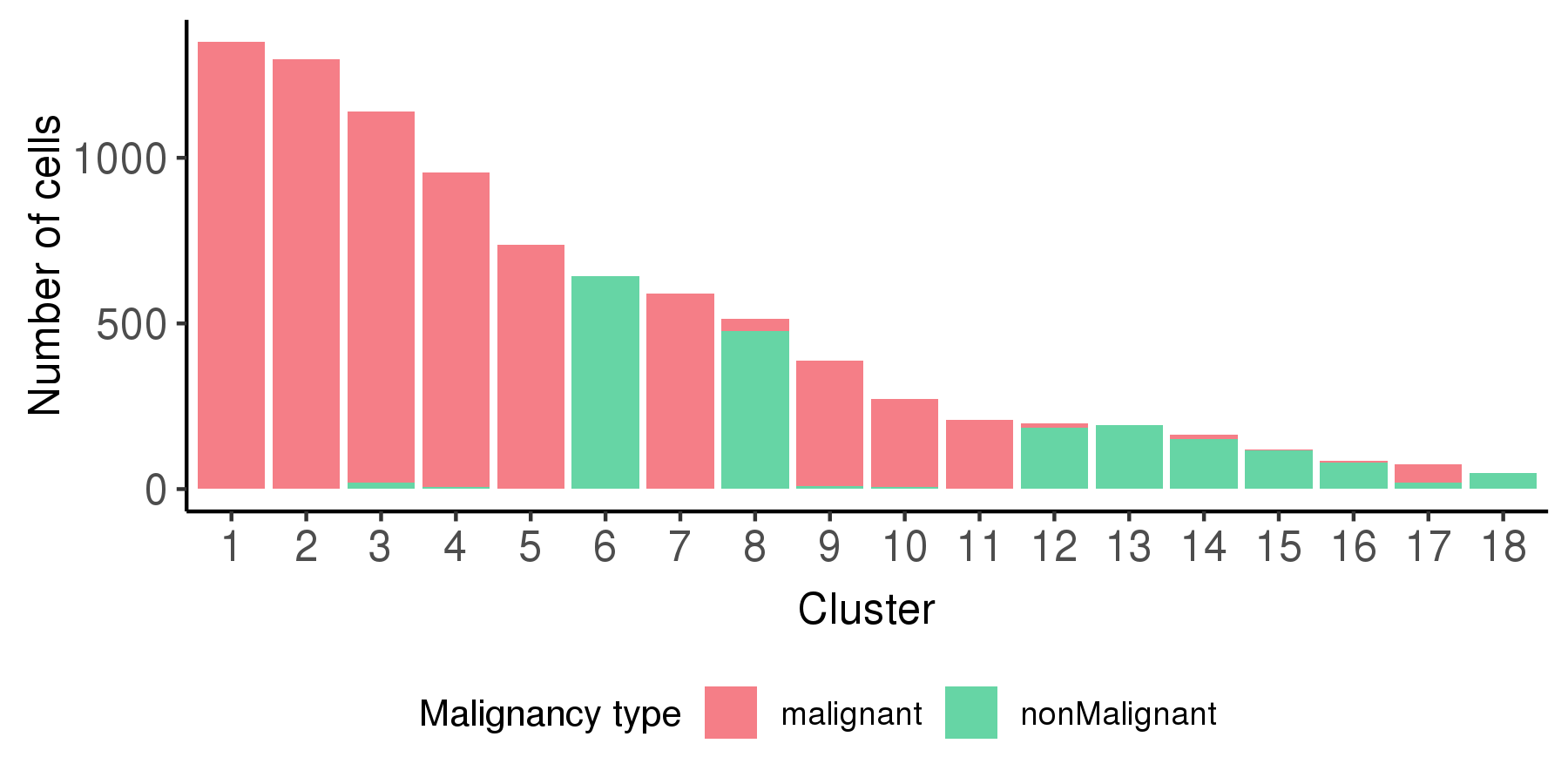
Following is a bar plot showing the relationship between cell cluster and cell malignancy type.
Following intra-tumor phenotypes and signatures heterogeneity analyses are mainly focused tumor cell identified before.
In runScAnnotation, the argument bool.intraTumor indicates whether to use the identified tumor clusters to perform following analyses.
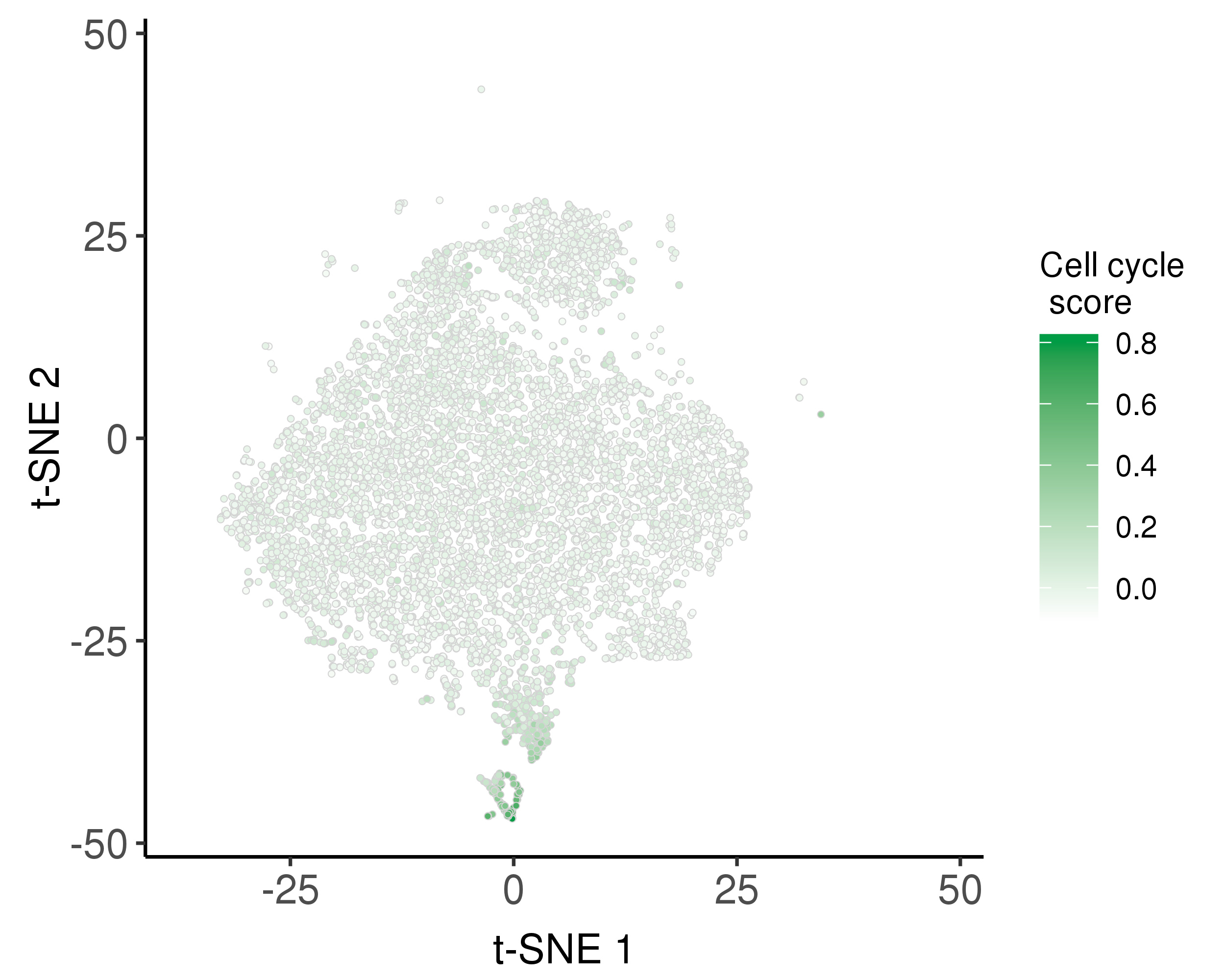
We used the Seurat AddModuleScore function to calculate the relative average expression of a list of G2/M and S phase markers as cell cycle scores.
In runScAnnotation, following arguments can determine detailed setting of this step.
-
bool.runCellCycleindicates whether to estimate cell cycle scores.
Following is the distribution of the estimated cell cycle scores.
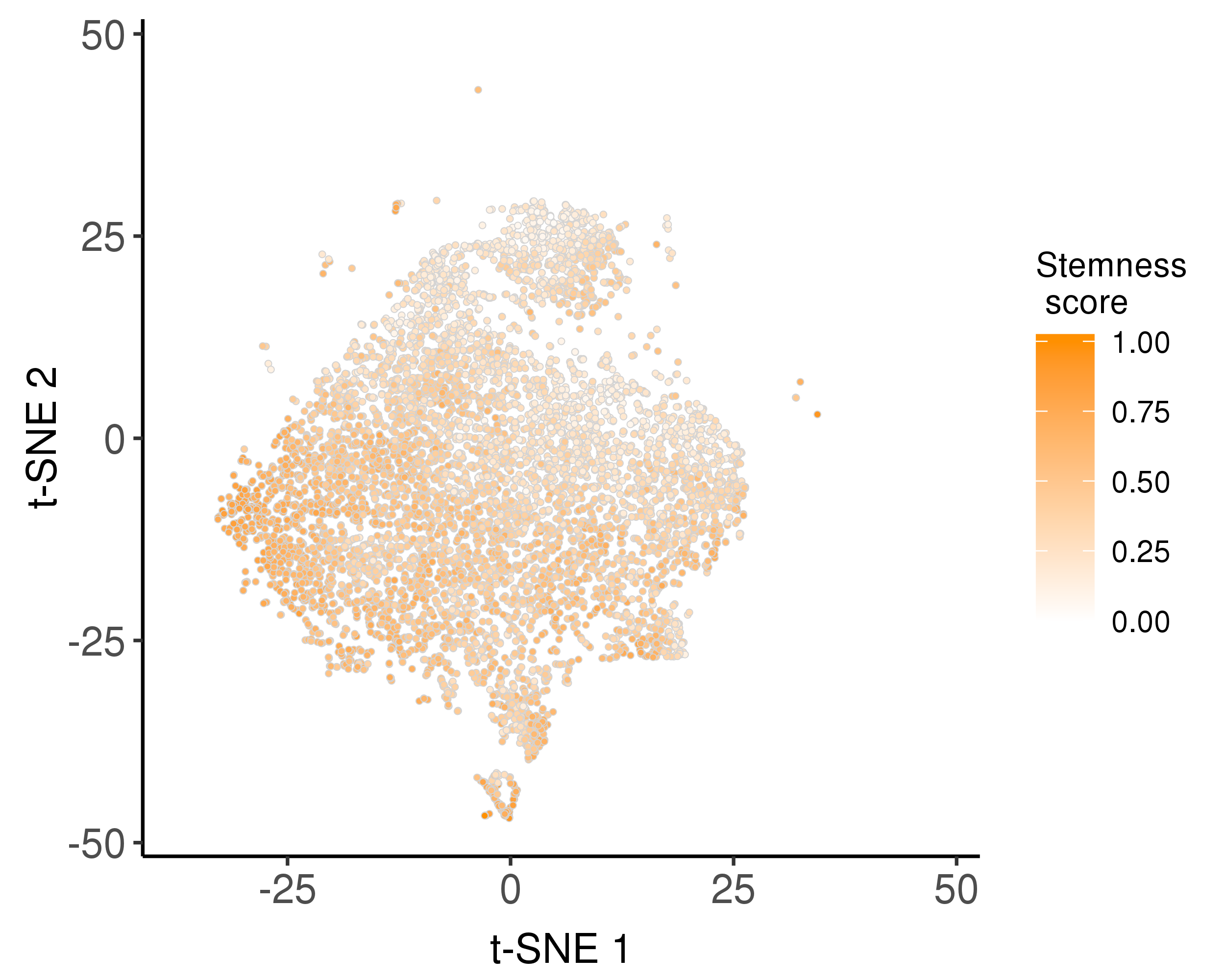
We trained a stemness signature by OCLR model and use it to estimate stemness scores.
In runScAnnotation, following arguments can determine detailed setting of this step.
-
bool.runStemnessindicates whether to estimate stemness scores.
Following is the distribution of the estimated stemness scores.
We provided two approaches to analyze known gene set
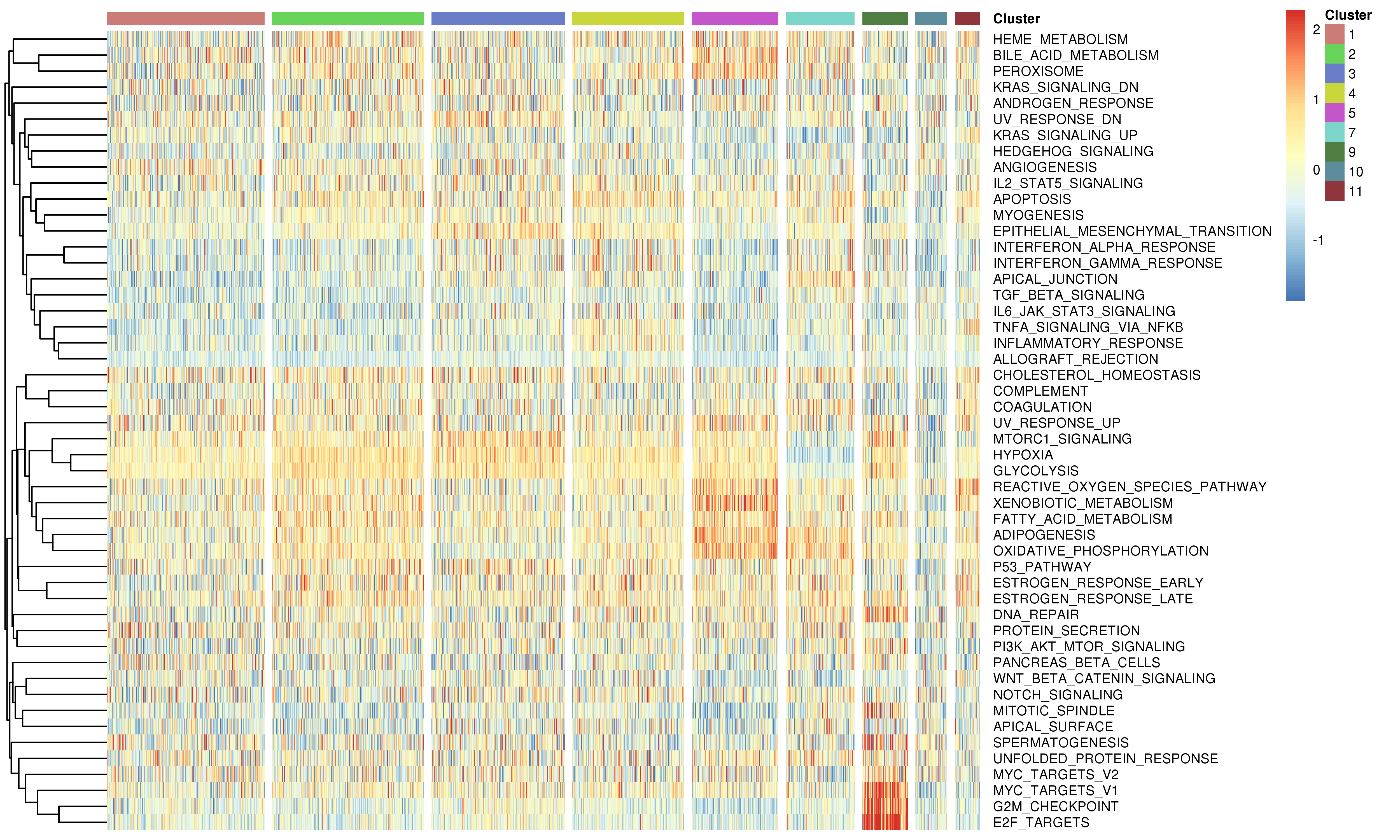
For the known gene set signature scores, such as pathways, We provided two approaches.
1.Use gene set variation analysis (GSVA) to estimate variation of the known gene set activities over the cells.
2.Use the relative average expression level across gene sets by using the Seurat AddModuleScore function.
By default, scCancer calculated signature scores of 50 hallmark gene sets from MSigDB and users can also input their own interested gene sets to the function.
In runScAnnotation, following arguments can determine detailed setting of this step.
-
bool.runGeneSetsindicates whether to estimate stemness scoresestimate gene sets signature scores. -
geneSetsindicates the gene sets to be analyzed. It should belistobject. The default isNULLand 50 hallmark gene sets from MSigDB will be used. -
geneSet.methodindicateds the method to be used in calculate gene set scores. Currently, onlyaverageandaverageare allowed.
Following is the example results of the gene set signature analysis.
We applied non-negative matrix factorization (NMF) to identify potential expression program signatures in unsupervised ways.
In runScAnnotation, following arguments can determine detailed setting of this step.
-
bool.runExprProgramindicates whether to run NMF to identify expression programs. -
nmf.rankindicates the decomposition rank used in NMF.
Following is the example results of the expression program signatures analysis.
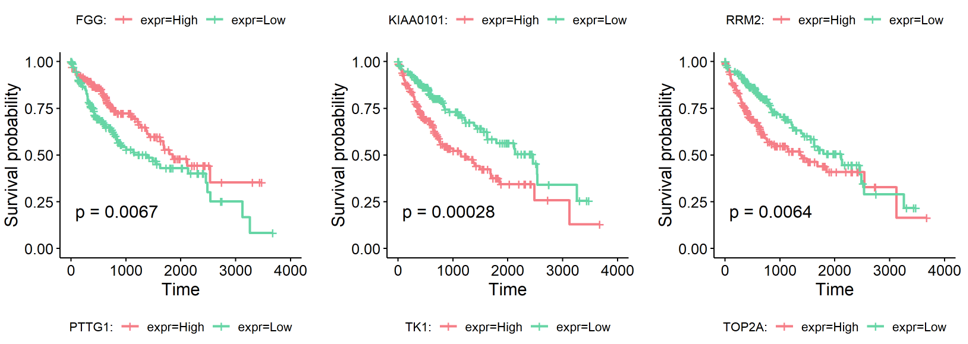
Based on the type-specific marker genes and gene signatures identified before, we provided an extra function runSurvival to perfrom survival analysis, which read the expression and survival data to plot survival curves and explore the relationship between genes or signatures expression levels and patient prognosis.
In runSurvival, following arguments can determine detailed setting of this step.
-
featuresindicates the names of marker genes or signatures to be analyzed. -
dataindicates the data used to perform survival analysis. It should be an expression or signature matrix with gene/signature by patient. The row names are the features' anmes. The columns are patients' labels. -
surv.timeindicates the survival time of patients. It should be in accord with the colimns indata. -
surv.eventindicates the status indicator of patients. 0=alive, 1=dead. It should be in accord with the colimns indata. -
cut.offindicates the percentage threshold to divide patients into two groups. The default is 0.5, which means the patients are divided by median. Other values, such as 0.4, means the first 40% patients are set "Low" group and the last 40% are set "High" group (the median 20% are discarded). -
savePathindicates the path to save the survival plots of genes/signatures. The default is NULL and the plots will be return without saving.
Following is the example results of survival analysis.
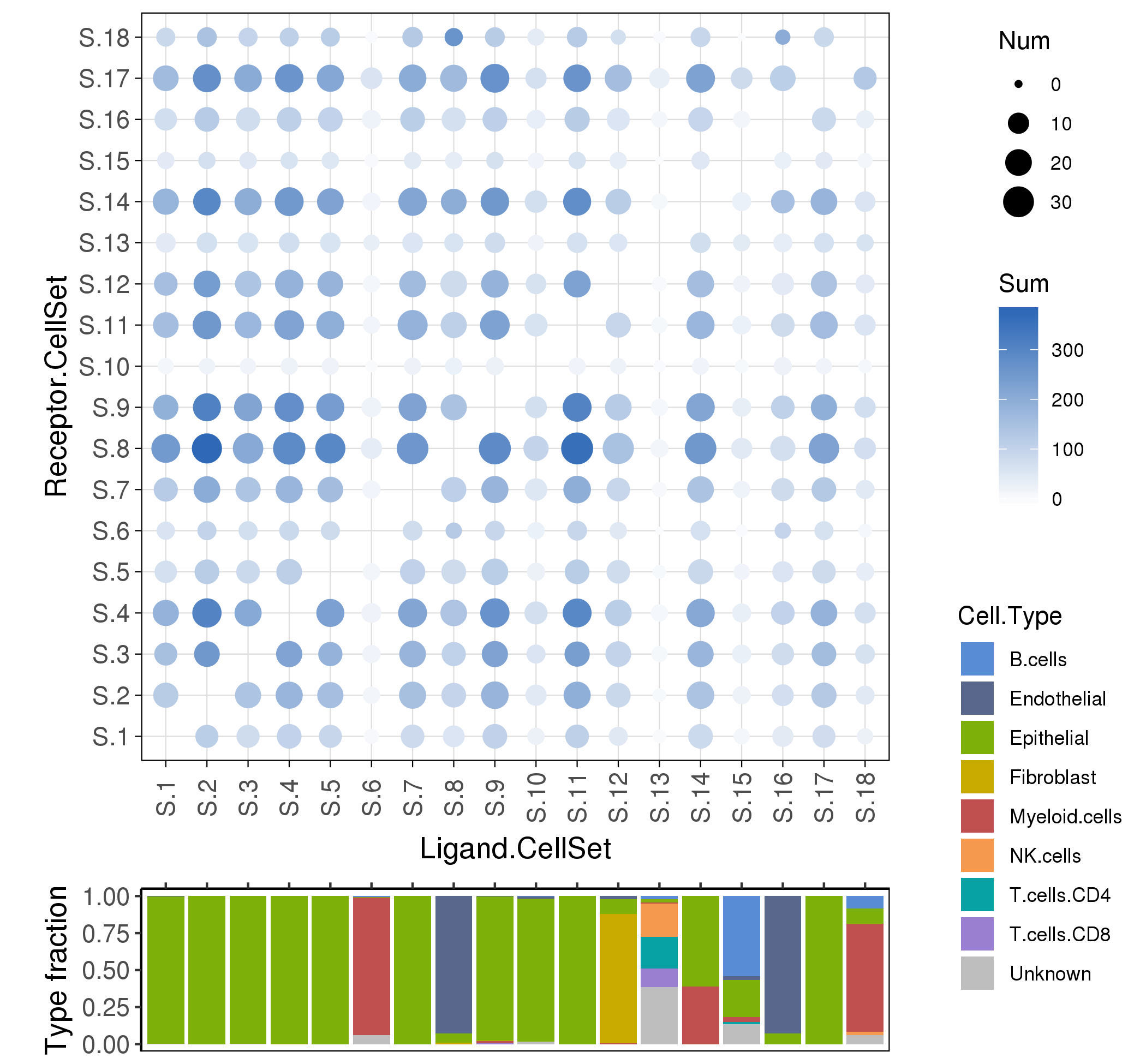
To analyze the ligand-receptor interactions between the various cell types in cancer micro-environment,
we used a ligand-receptor database FANTOM5, and estimate the interaction scores among cell sets (the default is clusters).
Following is the example results of cell interaction analysis.
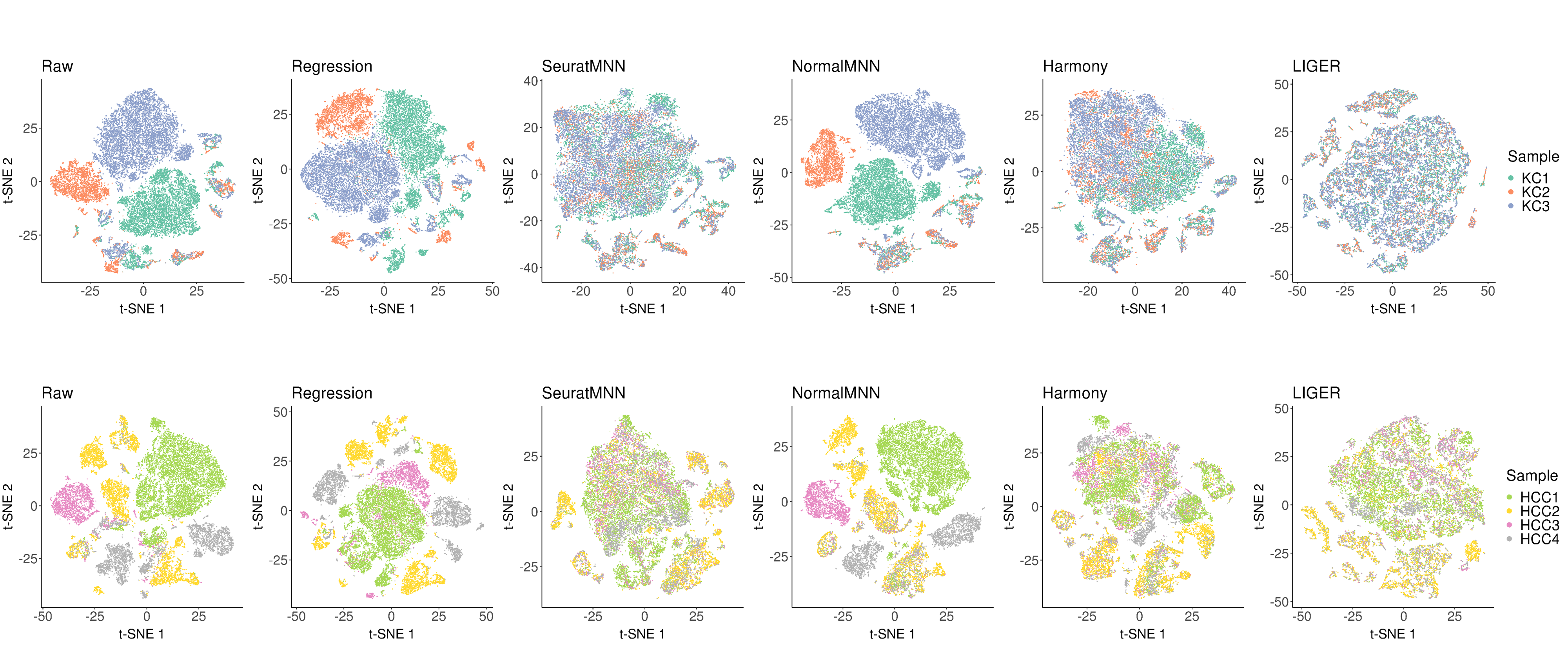
In scCancer, we provided six approaches to perform multi-sample integration analysis, which covered two of the most basic combination strategies (“Raw”, “Regression”), three best-performing algorithms after systematic evaluation (“SeuratMNN”, “Harmony”, “LIGER”), and a modified MNN version considering the inter-tumor heterogeneity (“NormalMNN”).
In runScCombination, following arguments can determine detailed setting of this step.
-
combNameindicates the label for the combined samples. -
comb.methodindicates the method to combine samples. The default is "NormalMNN". "Harmony", "NormalMNN", "SeuratMNN", "Raw", "Regression" and "LIGER" are optional. - Other arguments are similar to the single-sample module
runScAnnotation.
Following is the example results of the multi-sample data integration analysis.