Reseek is a protein structure search and alignment algorithm which improves sensitivity in protein homolog detection compared to state-of-the-art methods including DALI, TM-align and Foldseek with similar speed to Foldseek.
Reseek is based on sequence alignment where each residue in the protein backbone is represented by a letter in a novel “mega-alphabet” of 85,899,345,920 (∼1011) distinct states.
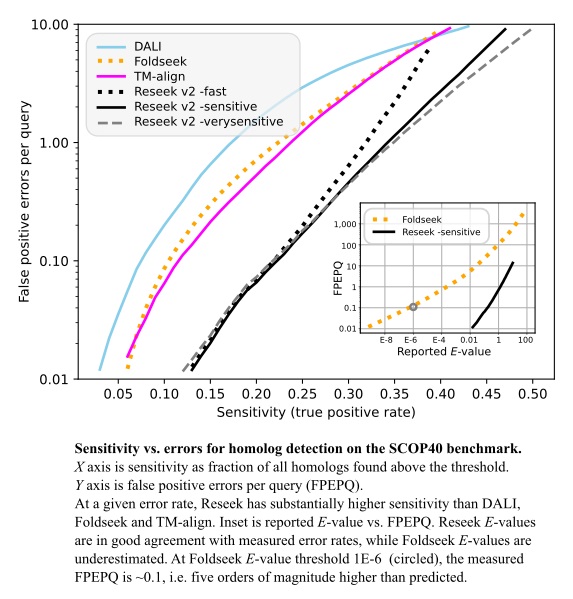
Method sensitivity was measured on the SCOP40 benchmark using superfamily as the truth standard, focusing on the regime with false-positive error rates <10 per query, corresponding to E<10 for an ideal E-value.

-search # Alignment (e.g. DB search, pairwise, all-vs-all)
-convert # Convert file formats (e.g. create DB)
-alignpair # Pair-wise alignment and superposition
Search against database
reseek -search STRUCTS -db STRUCTS -output hits.txt
# STRUCTS specifies structure(s), see below
Recommended format for large database is .bca, e.g.
reseek -convert /data/PDB_mirror/ -bca PDB.bca
Align and superpose two structures
reseek -alignpair 1XYZ.pdb -input2 2ABC.pdb
-aln FILE # Sequence alignment (text)
-output FILE # Rotated 1XYZ (PDB format)
All-vs-all alignment
reseek -search STRUCTS -output hits.txt
Output options for -search
-aln FILE # Alignments in human-readable format
-output FILE # Hits in tabbed text format
-columns name1+name2+name3...
# Output columns, names are
# query Query label
# target Target label
# qlo Start of aligment in query
# qhi End of aligment in query
# tlo Start of aligment in target
# thi End of aligment in target
# ql Query length
# tl Target length
# pctid Percent identity of alignment
# cigar CIGAR string
# evalue You can guess this one
# aq AQ (aln. qual., 0 to 1, >0.5 suggests homology)
# qrow Aligned query sequence with gaps (local)
# trow Aligned target sequence with gaps (local)
# qrowg Aligned query sequence with gaps (global)
# trowg Aligned target sequence with gaps (global)
# std query+target+qlo+qhi+ql+tlo+thi+tl+pctid+evalue
# default aq+query+target+evalue
Search and alignment options
-fast, -sensitive or -verysensitive # Required
-evalue E # Max E-value (default 10 unless -verysensitive)
-omega X # Omega accelerator (floating-point)
-minu U # K-mer accelerator (integer)
-gapopen X # Gap-open penalty (floating-point >= 0)
-gapext X # Gap-extend penalty (floating-point >= 0)
-dbsize D # DB size (nr. chains) for E-value (default actual size)
Convert between file formats
reseek -convert STRUCTS [one or more output options]
-cal FILENAME # .cal format, text with a.a. and C-alpha x,y,z
-bca FILENAME # .bca format, binary .cal, recommended for DBs
-fasta FILENAME # FASTA format
Create input for Muscle-3D multiple structure alignment:
reseek -pdb2mega STRUCTS -output structs.mega
STRUCTS argument is one of:
NAME.cif or NAME.mmcif # PDBx/mmCIF file
NAME.pdb # Legacy format PDB file
NAME.cal # C-alpha tabbed text format with chain(s)
NAME.bca # Binary C-alpha, recommended for larger DBs
NAME.files # Text file with one STRUCT per line,
# may be filename, directory or .files
DIRECTORYNAME # Directory (and its sub-directories) is searched
# for known file types including .pdb, .files etc.
Other options:
-log FILENAME # Log file with errors, warnings, time and memory.
-threads N # Number of threads, default number of CPU cores.
cd src/; chmod +x build_linux_x86.bash ; ./build_linux_x86.bash
cd src/ ; chmod +x build_osx_x86.bash ; ./build_osx_x86.bash
Load reseek.vcxproj into Microsoft Visual Studio and use the Build command.
Don't worry about a warning something like this, it's expected:
warning: Using 'dlopen' in statically linked applications requires at runtime the shared libraries from the glibc version used for linking
Edgar, Robert C. (2024) "Sequence alignment using large protein structure alphabets improves sensitivity to remote homologs" https://www.biorxiv.org/content/10.1101/2024.05.24.595840v2