{kind=link}
Judred is a peptide specific molecular descriptor generator, it is loosely based on the Mordred python package. The remit of this programme is to generate descriptors of peptides from only single-letter codes in order to provide a significantly faster method of generating descriptors for very large search spaces. It uses the HDF5 file format to store the data as this format is supported by many programming languages and platforms.
This programme is first used in the Journal of Chemical Theory and Computation article "Beyond tripeptides": https://doi.org/10.1021/acs.jctc.1c00159
Generate the full dataset of zwitterionic dipeptides:
julia judred.jl 2 Generate the full dataset of zwitterionic tripeptides:
julia judred.jl 3Generate the full dataset of zwitterionic tetrapeptides:
julia judred.jl 4 etc.
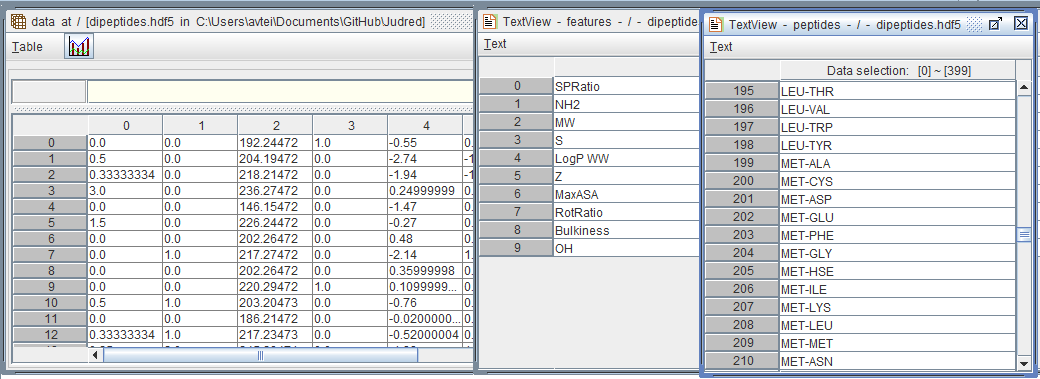
This image below shows example data (dipeptides) generated by the Judred programme.
The HDF5 database contain (left) a matrix of values contain the values of each descriptor for each peptide, a list of descriptor names (middle) and the list of peptides (right). These values can be recombined in for example a pandas DataFrame or the matrix of values used alone.
The unofficial version 2 (Judred_tiny.py) is written in Python and no longer uses the HDF5 file format. By removing the peptide labels and switching to the Apache Parquet format for data output we can reduce the dataset size by a factor of 13. This can be done as the specific peptide to which the parameters represent can be calculated from the parameters position in the dataset.
We have alse added the isoelectric point (pI) parameter as we found this to be a useful descriptor in an upcoming publication.
Since this is a complete dataset and all limits are know for each parameter is terms of min/min values we have 'cooked-in' scaling such that all values are saved between 0/-1 and 1 so there is no longer a need to scale after loading the dataset. As such the new filenaming scheme is: "tetrapeptides_normalized.parquet" instead of the old "tetrapeptides.hdf5".
Another improvement that has been made is the addition of GPU calculations. The arguments for the program are now:
python Judred_tiny.py [peptide_length] [chunk_size]
python Judred_tiny.py 4 10000Where the chunksize refers to how many values to calculate simulatneously (or as many as your processors will allow) - this in an important parameter particularly in the GPU computational as a lot of the 'processing' time is just moving data to and from the GPU so you want to set the chunck size to as large as your system will allow.
See the following on how this effects run time (10k/100k cpu/gpu):



