-
Notifications
You must be signed in to change notification settings - Fork 41
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
Merge pull request #95 from MDAnalysis/rel-19-post
release blog post for 0.19.0, 0.19.1, 0.19.2 close #94
- Loading branch information
Showing
1 changed file
with
128 additions
and
0 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,128 @@ | ||
| --- | ||
| layout: post | ||
| title: Release 0.19.0, 0.19.1, 0.19.2 | ||
| --- | ||
|
|
||
| A new version of MDAnalysis has been released! | ||
| Or rather, three new releases in quick succession, so we just talk about all of them in a single post. | ||
|
|
||
|
|
||
| This version brings a multitude of **fixes**, [**deprecations**](#deprecations), and **new features** including exciting additions from one of our two [2019 Google Summer of Code][] students (other cool new features from GSOC2018 will be unveiled in the next release...) and one [NSF REU][] student as well as **full Windows support**. | ||
| Some highlights are given below, whilst the [release notes][] list all the changes in this version. | ||
|
|
||
| ## GSoC 2018: Capped Distance Searches | ||
|
|
||
| GSoC student Ayush Suhane (@ayushsuhane) worked on integrating [faster distance search algorithms][] (such as the grid search in the new [`MDAnalysis.lib.nsgrid`](https://www.mdanalysis.org/docs/documentation_pages/lib/nsgrid.html) neighbor search library and [periodic KDTrees][]) for limited distance searches. His work improved many distance search based analysis methods like the calculation of radial distribution functions. There are now two new low-level functions [`MDAnalysis.lib.distances.capped_distance`](https://www.mdanalysis.org/docs/documentation_pages/lib/distances.html#MDAnalysis.lib.distances.capped_distance) and [`MDAnalysis.lib.distances.self_capped_distance`](https://www.mdanalysis.org/docs/documentation_pages/lib/distances.html#MDAnalysis.lib.distances.self_capped_distance), which find all pairwise distances between particles up to a given maximum distance. By specifying a maximum distance to the search, it is possible to optimize the search, leading to greatly improved performance especially in larger systems. | ||
|
|
||
| For example to find all contacts between oxygen and hydrogen up to 5.0 Å | ||
| ```python | ||
| from MDAnalysisTests.datafiles import PSF, DCD | ||
| import MDAnalysis as mda | ||
| from MDAnalysis.lib.distances import capped_distance | ||
|
|
||
| u = mda.Universe(PSF, DCD) | ||
|
|
||
| oxy = u.select_atoms('name O*') | ||
| hyd = u.select_atoms('name H*') | ||
|
|
||
| idx, dists = capped_distance(oxy.positions, hyd.positions, box=u.dimensions, max_cutoff=5.0) | ||
| ``` | ||
| Unlike [`distance_array`](https://www.mdanalysis.org/docs/documentation_pages/lib/distances.html#MDAnalysis.lib.distances.distance_array), | ||
| which returns a matrix of every pairwise distance, | ||
| here a sparse representation is returned, | ||
| where `idx` is a `(n, 2)` array of the indices of the atoms | ||
| and `dists` is the distance between these atoms. | ||
|
|
||
| For full details on the implementation of this and the expected performance improvements | ||
| see Ayush's post on [faster distance search algorithms][]. | ||
|
|
||
| ## Analysis improvements | ||
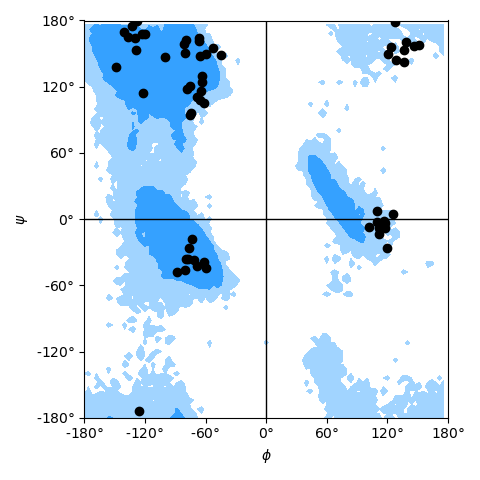
| **NSF REU** student [Henry Mull][] implemented a new analysis module [`MDAnalysis.analysis.dihedrals`][] which includes analysis classes for | ||
| - **fast dihedral angle calculation** (useful for featurization and dimensionality reduction in dihedral space as well as conformational analysis), | ||
| - **Ramachandran** analysis (protein backbone dihedrals) including functions and data to plot allowed and generously allowed regions, | ||
| - **Janin** analysis (protein sidechain dihedrals) | ||
|
|
||
|  | ||
|
|
||
|
|
||
| [Irfan Alibay][] improved the [`density_from_Universe()`](https://www.mdanalysis.org/docs/documentation_pages/analysis/density.html#MDAnalysis.analysis.density.density_from_Universe) function, which now allows the user to exactly specify the region in which a density should be calculated. | ||
| In this way, it becomes easier to calculate densities on identical grids for different simulations so that these densities can be compared more easily. | ||
|
|
||
| [Shujie Fan][] added [site specific radial distribution function][] analysis. The [`InterRDF_s()`][] function calculates the radial distribution function relative to a single or a few particles (a "site"). | ||
| The function helps with the analysis of the coordination of ions and ligands in binding sites of proteins or other biomolecules, for instance, the distribution of oxygen ligand atoms around sodium ions. | ||
| Importantly, many of these sites can be computed at the same time, which improves performance because the most time consuming part of almost all analysis tasks, the loading of the trajectory data from disk into memory, only has to be done once, and then multiple computations can be performed with the data in memory. | ||
|
|
||
| ## Windows support | ||
| Since 0.19.2, Windows is fully supported *under Python 3.4+* (Python 2.7 is not officially supported because of technical difficulties, which we decided not to address because of limited developer time and the fact that Python 3 is now the recommended version of Python). | ||
|
|
||
| For Windows we recommend the [conda installation][]. | ||
| If you want to install with `pip` or from source then you will need the full Microsoft developer environment with Microsoft Visual C++ 14.0. | ||
|
|
||
| ## Improvements to file readers | ||
|
|
||
| - For the LAMMPS dump reader one can now pass an [`atom_style` keyword][atom_style] argument to specify what is on each line, for instance, `atom_style = 'id type charge x y z'`. This provides much greater flexibility to read different dump files. | ||
| - The [ChainReader][] with argument `continuous=True` can now correctly handle continuous XTC or TRR trajectories that were split into multiple files with Gromacs `gmx mdrun -noappend`; in particular it will handle overlapping frames in such a way that only the last generated frame is included and the trajectory contains strictly monotonously increasing time steps. This means, Gromacs folks will never have to manually concatenate their trajectories for use with MDAnalysis. | ||
| - When slicing a trajectory as with `traj = u.trajectory[start:stop:step]` then this returns now a trajectory slice object (an iterable), which can be passed around and then iterated over: essentially, it's a trajectory that knows that it should only deliver a subset of frames. Functions like `len(traj)` are also fast (O(1)), very similar to how Python 3's `range()` object functions. See [FrameIterators][] for more details. | ||
| - The [MemoryReader][] is now a full first-class citizen in MDAnalysis, which can keep coordinates *and* velocities, forces, box dimensions in memory. It is a versatile swiss-army knife, both for fast analysis and also visualization with nglview (e.g., [fit trajectory in memory, then visualize][trajectory_magic.ipynb]). | ||
| - new hybrid36 PDB-like format | ||
| - The Gromacs TPR parser now reads SETTLE constraints. | ||
| - The Amber top parser reads bonds/angles/dihedrals. | ||
|
|
||
|
|
||
| ## Miscellaneous performance improvements | ||
|
|
||
| - `guess_bonds` (thanks to above methods) | ||
| - geometry selections faster (thanks to above) | ||
| - `make_whole` (C++ rewrite) | ||
| - fragment finding (C++ rewrite) | ||
| - improvements to AtomGroup internals | ||
|
|
||
| ## Deprecations | ||
|
|
||
| This release brings a few new deprecations as the package heads towards a final API: | ||
|
|
||
| - `start`/`stop`/`step` are deprecated in the initialization of Analysis classes. These parameters should instead be given to the run() method of the class. | ||
| - Almost all "`save()`", "`save_results()`", "`save_table()`" methods in analysis classes. | ||
| - Deprecated use of `core.flags`, the deprecation messages for this give advice | ||
| on how to replace this functionality. | ||
| - Default ``filename`` directory of `align.AlignTraj` is deprecated and will change in 1.0 to the current directory. | ||
|
|
||
| ## Author statistics | ||
|
|
||
| Altogether this represents the work of 16 contributors from around the world, | ||
| and featured the work of five new contributors: | ||
| - [Irfan Alibay][] | ||
| - [Shujie Fan][] | ||
| - [Andrew R. McCluskey][] | ||
| - [Henry Mull][] | ||
| - [Paul Smith][] | ||
|
|
||
|
|
||
| ## Upgrading to MDAnalysis version 0.19.x | ||
|
|
||
| To get all these features run either `conda update -c conda-forge mdanalysis` | ||
| or `pip install --upgrade MDAnalysis`. | ||
|
|
||
| — The MDAnalysis Team | ||
|
|
||
| [2019 Google Summer of Code]: {{ site.baseurl }}{% post_url 2018-04-26-gsoc-students %} | ||
| [NSF REU]: https://becksteinlab.physics.asu.edu/positions/115/research-experience-for-undergraduates-summer-2018 | ||
| [release notes]: {{ site.github.wiki }}/ReleaseNotes0190 | ||
| [periodic KDTrees]: https://www.mdanalysis.org/mdanalysis/documentation_pages/lib/pkdtree.html | ||
| [faster distance search algorithms]: {{ site.baseurl }}{% post_url 2018-11-28-gsoc18-distancesearch %} | ||
| [visualization of trajectories with nglview]: {{ site.baseurl }}{% post_url 2016-03-14-nglview %} | ||
| [`MDAnalysis.analysis.dihedrals`]: https://www.mdanalysis.org/docs/documentation_pages/analysis/dihedrals.html | ||
| [site specific radial distribution function]: https://www.mdanalysis.org/docs/documentation_pages/analysis/rdf.html#site-specific-radial-distribution-function | ||
| [`InterRDF_s()`]: https://www.mdanalysis.org/docs/documentation_pages/analysis/rdf.html#MDAnalysis.analysis.rdf.InterRDF_s | ||
| [conda installation]: https://www.mdanalysis.org/pages/installation_quick_start/#conda | ||
| [atom_style]: https://www.mdanalysis.org/docs/documentation_pages/topology/LAMMPSParser.html#atom-styles | ||
| [FrameIterators]: https://www.mdanalysis.org/docs/documentation_pages/coordinates/base.html#frameiterators | ||
| [ChainReader]: https://www.mdanalysis.org/docs/documentation_pages/coordinates/chain.html | ||
| [MemoryReader]: https://www.mdanalysis.org/docs/documentation_pages/coordinates/memory.html | ||
| [trajectory_magic.ipynb]: https://github.com/MDAnalysis/WorkshopHackathon2018/blob/master/01_IntroToMDAnalysis/notebooks/trajectory_magic.ipynb | ||
| [Irfan Alibay]: https://github.com/IAlibay | ||
| [Shujie Fan]: https://github.com/VOD555 | ||
| [Andrew R. McCluskey]: https://github.com/arm61 | ||
| [Henry Mull]: https://github.com/hfmull | ||
| [Paul Smith]: https://github.com/p-j-smith | ||
|
|