

Resistify is a program which rapidly identifies and classifies plant resistance genes from protein sequences. It is designed to be lightweight and easy to use.

Resistify is available via the Bioconda channel:
conda create -n resistify bioconda::resistify
conda activate resistify
Note
If you want to use the GPU-accelerated pipelines, conda may fail to install a GPU-ready version of pytorch. If this occurs, try installing pytorch-gpu bioconda::resistify instead.
When using conda, please ensure that your Bioconda has been configured correctly.
Docker/Podman containers are also available through the biocontainers repository.
To use these with - for example - singularity, simply run:
singularity exec docker://quay.io/biocontainers/resistify:<tag-goes-here>
If you are having issues with conda, you can instead try installing directly from the repository:
pip install https://github.com/SwiftSeal/resistify/archive/refs/tags/v0.6.2.tar.gzNote that resistify requires hmmer to be installed and available in your system's PATH, which will not be installed automatically when using pip.
To predict NLRs within a set of protein sequences, simply run:
resistify nlr <input.fa>
and Resistify will identify and classify NLRs, and return some files:
results.tsv- A table containing the primary results ofResistify.motifs.tsv- A table of all the NLR motifs identified for each sequence.domains.tsv- A table of all the domains identified for each sequence.annotations.tsv- A table of the raw annotations for each sequence.nbarc.fasta- A fasta file of all the NB-ARC domains identified.nlr.fasta- A fasta file of all NLRs identified.
By default, Resistify will only return sequences with NB-ARC domains.
If you wish to identify highly fragmented NLRs, you can use the --retain option which will predict and report NLR-associated motifs for all sequences.
It'll be a bit slower!
If you want to increase the sensitivity of coiled-coil domain annotation, you can use the option --coconat.
This will use CoCoNat to predict coiled-coil domains.
In practice, I wouldn't expect this mode to pick up on a significant number of missed CC domains, but it can pick up on cryptic CCs that do not have an identifiable EDVID motif.
Resistify carries out an initial search for common NLR domains to quickly filter and annotate the input sequences.
Then, Resistify executes a re-implementation of NLRexpress to conduct a highly accurate search for NLR-associated motifs.
If --coconat is used, this will also be executed to scavenge for potentially missed coiled-coil domains.
Together, this evidence is used to classify NLRs according to their domain architecture.
Important
This pipeline is currently in development - due to other commitments I can't currently benchmark this properly and make no guarantees to its accuracy yet! Feedback is appreciated.
To predict PRRs within a set of protein sequences, simply run:
resistify prr <input.fa>
and Resistify will identify and classify PRRs, and return some files:
results.tsv- A table containing the primary results ofResistify.motifs.tsv- A table of all the LRR motifs identified for each sequence.domains.tsv- A table of all the domains identified for each sequence.annotations.tsv- A table of the raw annotations for each sequence.prr.fasta- A fasta file of all PRRs identified.
Warning
This pipeline is GPU-accelerated and will be extremely slow on CPU only.
First, Resistify searches for domains associated with a recently described classification system for RLP/RLKs.
Then, a re-implementation of TMbed is used to predict transmembrane domains - sequences with a single α-helix transmembrane domain and an extracellular domain of at least 50 amino acids are considered as RLPs.
Finally, NLRexpress is used to to identify LRR domains.
Sequences are classified as being either RLPs or RLKs depending on the presence of an internal kinase domain, and are classified according to their extracellular domain.
Note
This only applies to the --coconat and PRR pipelines! The standard NLR pipeline does not require any external databases.
By default, Resistify will automatically download the models required for CoConat and TMbed to your $HOME/.cache directory.
If you'd like to manually install the databases instead, you can use the resistify download_models utility to download these to a directory of your choice.
To provide these local models to the CoCoNat and TMbed processes, simply pass the path of the models directory via the --models argument.
Approximately 13G of disk space is required.
| Sequence | Length | Motifs | Domains | Classification | NBARC_motifs | MADA | MADAL | CJID |
|---|---|---|---|---|---|---|---|---|
| ZAR1 | 852 | CNNNNNNNNNLLLLLLLLLL | mCNL | CNL | 9 | False | True | False |
The main column of interest is "Classification", where we can see that it has been identified as a canonical CNL. The "Motifs" column indicates the series of NLR-associated motifs identified across the sequence - this can be useful if an NLR has an undetermined or unexpected classification. The columns "MADA", "MADAL", and "CJID" correspond to common NLR sequence signatures. Here, it appears that ZAR1 has a MADA-like motif.
| Sequence | Length | Type | Classification | Signal_peptide |
|---|---|---|---|---|
| fls2 | 1174 | RLK | LRR | True |
For PRRs, sequences can be of the type RLP or RLK - both are single pass transmembrane proteins, and RLKs have an internal kinase domain. Classification refers to the domains identified in the external region. If multiple domains are identified, they will each be reported as a semi-colon separated list. If a signal peptide is identified in the sequence, this is reported accordingly.
| Sequence | Motif | Position | Probability | Downstream_sequence | Motif_sequence | Upstream_sequence |
|---|---|---|---|---|---|---|
| ZAR1 | extEDVID | 65 | 0.9974 | LVADL | RELVYEAEDILV | DCQLA |
| ZAR1 | VG | 159 | 0.9924 | YDHTQ | VVGLE | GDKRK |
| ZAR1 | P-loop | 188 | 1.0 | IMAFV | GMGGLGKTT | IAQEV |
| ZAR1 | RNSB-A | 211 | 0.9981 | EIEHR | FERRIWVSVS | QTFTE |
| ZAR1 | Walker-B | 259 | 0.973 | QYLLG | KRYLIVMD | DVWDK |
| ZAR1 | RNSB-B | 290 | 0.9846 | RGQGG | SVIVTTR | SESVA |
| ZAR1 | RNSB-C | 317 | 0.9994 | HRPEL | LSPDNSWLLF | CNVAF |
| ZAR1 | RNSB-D | 417 | 0.9875 | SHLKS | CILTLSLYP | EDCVI |
| ZAR1 | GLPL | 356 | 0.9998 | VTKCK | GLPLT | IKAVG |
| ZAR1 | MHD | 486 | 0.9965 | IITCK | IHD | MVRDL |
| ZAR1 | LxxLxL | 511 | 0.9398 | PEGLN | CRHLGI | SGNFD |
| ZAR1 | LxxLxL | 560 | 0.9973 | TDCKY | LRVLDI | SKSIF |
| ZAR1 | LxxLxL | 587 | 0.9993 | ASLQH | LACLSL | SNTHP |
| ZAR1 | LxxLxL | 611 | 0.9995 | EDLHN | LQILDA | SYCQN |
| ZAR1 | LxxLxL | 635 | 0.999 | VLFKK | LLVLDM | TNCGS |
| ZAR1 | LxxLxL | 685 | 0.9987 | KNLTN | LRKLGL | SLTRG |
| ZAR1 | LxxLxL | 712 | 0.9723 | INLSK | LMSISI | NCYDS |
| ZAR1 | LxxLxL | 740 | 0.9995 | TPPHQ | LHELSL | QFYPG |
| ZAR1 | LxxLxL | 765 | 0.9976 | HKLPM | LRYMSI | CSGNL |
| ZAR1 | LxxLxL | 817 | 0.9391 | QSMPY | LRTVTA | NWCPE |
Here, the positions, probabilities, and sequence of NLRexpress motif hits are listed. The five amino acids upstream and downstream of the motif site are also provided. In PRR mode, only LRR motifs will be reported.
| Sequence | Domain | Start | End |
|---|---|---|---|
| ZAR1 | MADA | 0 | 21 |
| ZAR1 | CC | 4 | 129 |
| ZAR1 | NB-ARC | 162 | 410 |
| ZAR1 | LRR | 511 | 817 |
This file contains the coordinates of the domains identified by Resistify.
| Sequence | Domain | Start | End | E_value | Score | Source |
|---|---|---|---|---|---|---|
| ZAR1 | MADA | 0 | 21 | 1.5e-06 | 16.2 | HMM |
| ZAR1 | CC | 4 | 128 | 2.3e-23 | 70.0 | HMM |
| ZAR1 | CC | 27 | 48 | NA | NA | Coconat |
| ZAR1 | CC | 60 | 75 | NA | NA | Coconat |
| ZAR1 | CC | 113 | 129 | NA | NA | Coconat |
| ZAR1 | NB-ARC | 162 | 410 | 1.4e-89 | 287.2 | HMM |
| ZAR1 | LRR | 511 | 817 | NA | NA | NLRexpress |
This file contains the raw annotations for each sequence, and the method which was used to identify them.
I've kept the output files of Resistify fairly minimal so that users can carry out their own analysis/visualisation.
Here are some examples of how Resistify can be used to create basic plots.
Resistify extracts the NB-ARC domains of each hit so we can easily build a phylogenetic tree.
Here, we create a tree rooted on the NB-ARC domain of CED-4.
The mafft | fastree method is used here for brevity rather than accuracy.
echo -e ">ced4\nREYHVDRVIKKLDEMCDLDSFFLFLHGRAGSGKSVIASQALSKSDQLIGINYDSIVWLKDSGTAPKSTFDLFTDILLMLARVVSDTDDSHSITDFINRVLSRSEDDLLNFPSVEHVTSVVLKRMICNALIDRPNTLFVFDDVVQEETIRWAQELRLRCLVTTRDVEISNAASQTCEFIEVTSLEIDECYDFLEAYGMPMPVGEKEEDVLNKTIELSSGNPATLMMFFKSCEPKTFEKMAQLNNKLESRGLVGVECITPYSYKSLAMALQRCVEVLSDEDRSALAFAVVMPPGVDIPVKLWSCVIPVD" >> output/nbarc.fasta
mafft output/nbarc.fasta | fasttree > output/nbarc.tree
We can now plot the tree:
library(tidyverse)
library(ggtree)
tree <- read.tree("output/nbarc.tree")
tree <- treeio::root(tree, outgroup = "ced4")
results <- read_tsv("output/results.tsv") |>
mutate(Sequence = paste0(Sequence, "_1"))
myplot <- ggtree(tree, layout = "circular") %<+% results
myplot <- myplot +
geom_tippoint(aes(colour = Classification))

Sometimes, it might be of interest to plot the distribution of domains and motifs across each NLR.
Achieving this with Resistify is quite simple:
library(tidyverse)
motif_translation = c(
"extEDVID" = "CC",
"bA" = "TIR",
"aA" = "TIR",
"bC" = "TIR",
"aC" = "TIR",
"bDaD1" = "TIR",
"aD3" = "TIR",
"VG" = "NB-ARC",
"P-loop" = "NB-ARC",
"RNSB-A" = "NB-ARC",
"Walker-B" = "NB-ARC",
"RNSB-B" = "NB-ARC",
"RNSB-C" = "NB-ARC",
"RNSB-D" = "NB-ARC",
"GLPL" = "NB-ARC",
"MHD" = "NB-ARC",
"LxxLxL" = "LRR"
)
domains <- read_tsv("output/domains.tsv")
results <- read_tsv("output/results.tsv")
motifs <- read_tsv("output/motifs.tsv") |>
mutate(Domain = motif_translation[Motif])
myplot <- ggplot() +
geom_segment(data = results, aes(y = Sequence, yend = Sequence, x = 0, xend = Length)) +
geom_segment(data = domains, aes(y = Sequence, yend = Sequence, x = Start, xend = End, colour = Domain)) +
geom_point(data = motifs, aes(y = Sequence, x = Position, colour = Domain))

Cute! NB: Some false-positive motif hits are evident in this example - it might be of interest to not plot them, or plot only LRR motifs which tend to be a bit more informative.
Q: Can Resistify be used to predict resistance genes from genomic data?
A: Unfortunately, Resistify cannot be directly applied to a genome to predict resistance genes, unlike tools such as NLR-Annotator.
If gene annotations are unavailable for your genome, my advice would be to use a tool like Helixer to perform ab initio gene prediction.
Q: According to the Motif string, some of my genes have NLR motifs in unexpected places - are these significant?
A: False positives do occur for the motif predictions, and unexpected predictions such as a single CC motif in the LRR domain are unlikely to be representative of a true domain annotation. You can find a figure of the prediction accuracy rates for each predictor here. False positives shouldn't interfere with the classification accuracy.
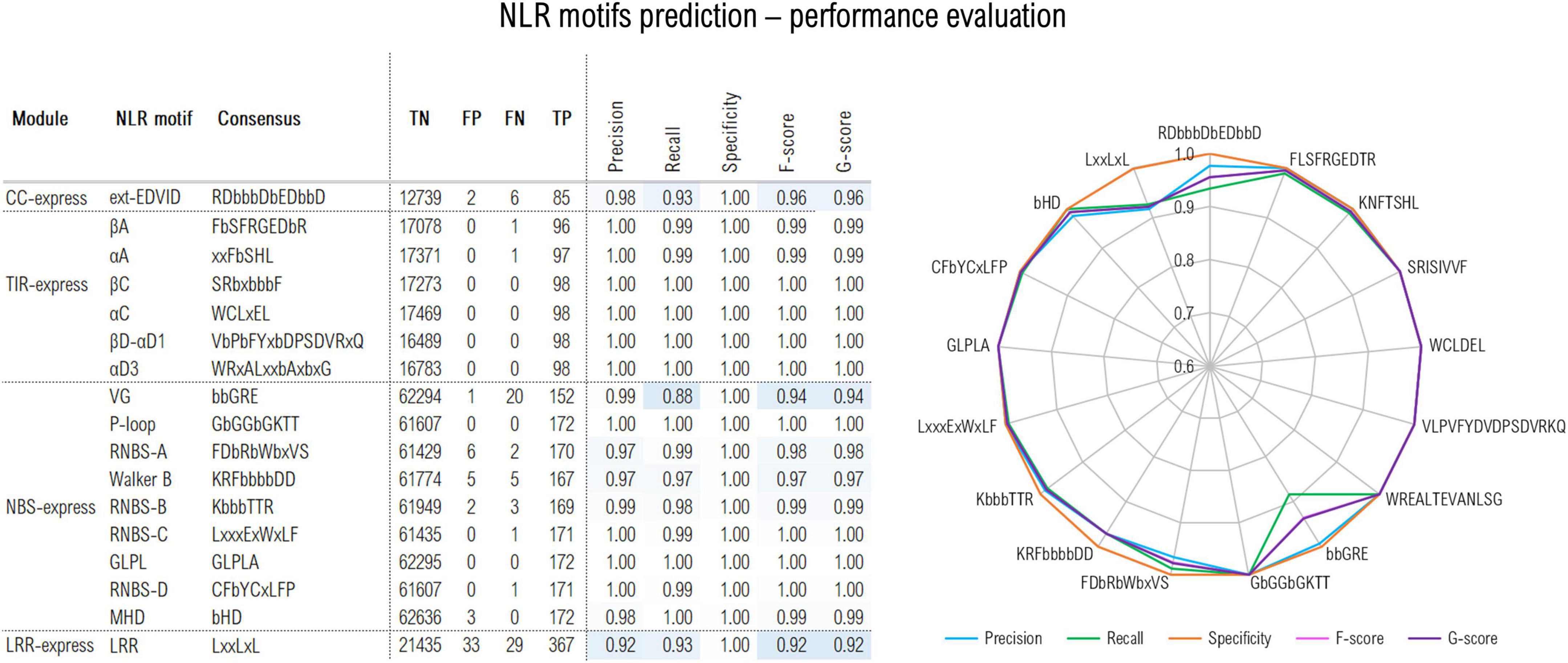{kind=link}
Q: The NLRexpress step is quite slow - how can I speed it up?
A: More threads!
The process is relatively fast on a non-NLR sequence, but can be quite slow when applied to an NLR.
Resistify will automatically use as many threads as possible - I've used up to 128 threads and it scales fairly well.
It's primarily due to the underlying jackhmmer process, which is slow when applied to NLRs, but not non-NLRs.
As a result the --retain option doesn't have as much of a performance impact as you might expect.
Contributions are greatly appreciated! If you experience any issues running Resistify, please get in touch via the Issues page. If you have any suggestions for additional features, get in touch!
Smith, M., Jones, J. T., & Hein, I. (2024). Resistify - A rapid and accurate annotation tool to identify NLRs and study their genomic organisation. bioRxiv, 2024-02. https://doi.org/10.1101/2024.02.14.580321
You must also cite:
Martin, E. C., Spiridon, L., Goverse, A., & Petrescu, A. J. (2022). NLRexpress—A bundle of machine learning motif predictors—Reveals motif stability underlying plant Nod-like receptors diversity. Frontiers in Plant Science, 13, 975888. https://doi.org/10.3389/fpls.2022.975888
If you use the CoCoNat module, please cite:
Madeo, G., Savojardo, C., Manfredi, M., Martelli, P. L., & Casadio, R. (2023). CoCoNat: a novel method based on deep learning for coiled-coil prediction. Bioinformatics, 39(8), btad495. https://doi.org/10.1093/bioinformatics/btad495
If you use the PRR module, please cite:
Bernhofer, M., & Rost, B. (2022). TMbed: transmembrane proteins predicted through language model embeddings. BMC bioinformatics, 23(1), 326. https://doi.org/10.1186/s12859-022-04873-x