Home
Welcome to the HybPiper wiki! These documents will cover more in depth installation instructions and tutorials for different ways to use HybPiper.
Troubleshooting: Common Issues
HybPiper expects "clean" sequencing reads in FASTQ format. What "clean" means is up to the user, but we recommend Trimmomatic, which can remove poor quality reads, trim poor quality base calls, and remove adapter sequences. If you are interested in calculating coverage depth exactly, you may also want to remove PCR duplicates, but this is not necessary.
HybPiper can accept either single-end or paired-end data. As of version 1.2, you may also include a file containing unpaired reads with the --target flag.
It is a good idea to rename your read-files so that they contain a unique identifier for each sample. This will be useful for running HybPiper automatically on many samples. For an example, see the test dataset in the Tutorial.
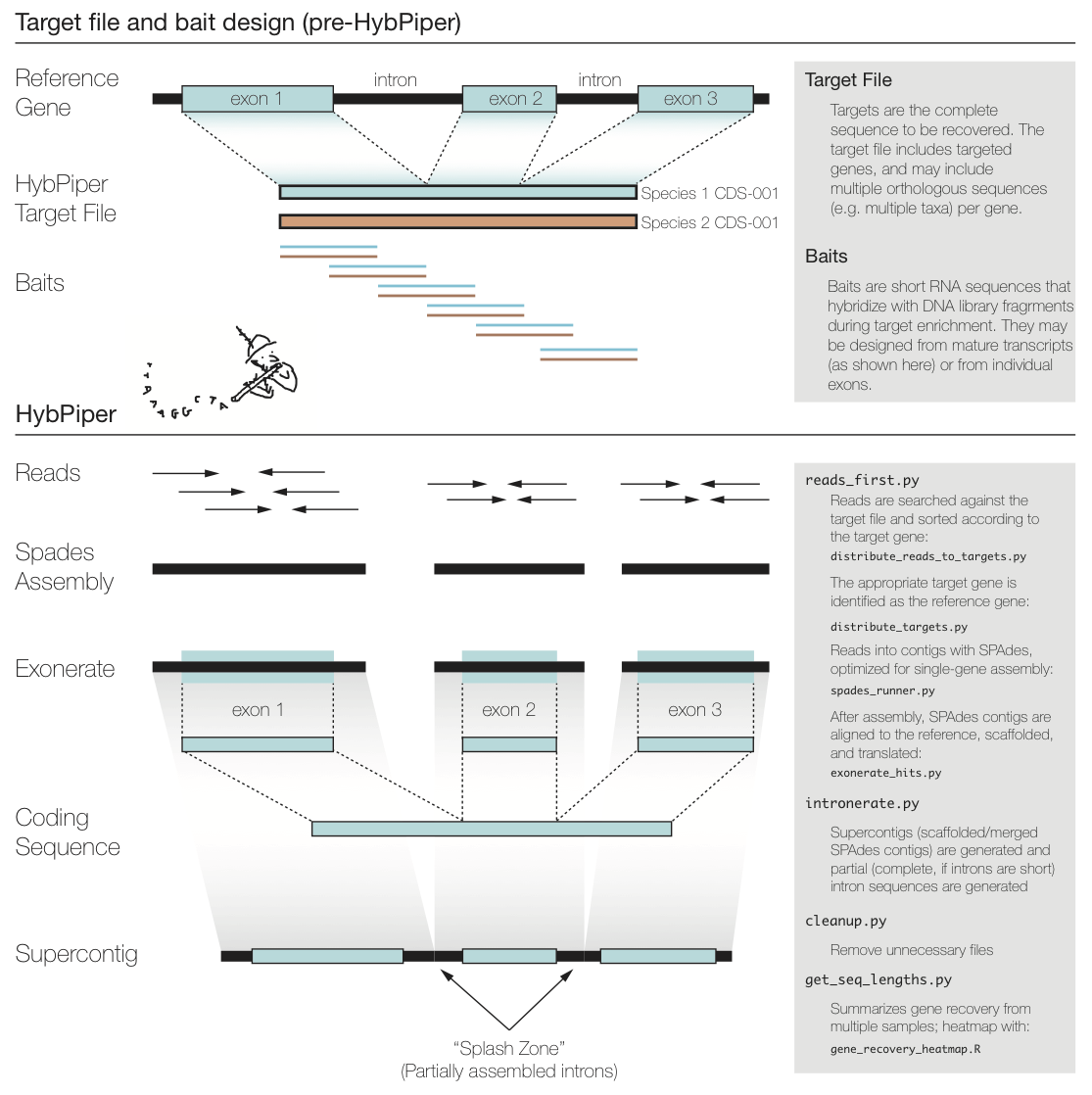
The figure above summarizes a typical exon enrichment setup that targets coding regions of genes for phylogenetics. The baits (short lines) are tiled across the exons. HybPiper uses the coding sequence portion of a gene for both mapping sequencing reads and for alignment of contigs. When generating a "target file" for HybPiper (see below), exon sequences should be concatenated for each gene. Another way to think of the target sequence is as the mature RNA sequence produced by this gene.
The reason HybPiper treats coding sequences this way is to more accurately detect the exon/intron boundary when aligning the assembled contigs against the reference sequence. HybPiper will align assembled contigs to the full gene sequence and will separate exon sequences from intron sequences.
HybPiper uses target sequences to first sort the reads and then extract exon sequences from assembled contigs, for each gene sepearately. In order to run HybPiper, you will need to prepare a "target file," a FASTA file that contains one or more homologous sequences for each gene.
If you are using HybPiper on data generated using HybSeq, the target file should contain the full-length reference sequences for each gene, NOT the probe sequences.
IMPORTANT: If you are using BLAST to map reads to targets, you need a PROTEIN target file. If you are using BWA, you need a NUCLEOTIDE target file.
Construct a "target file" of protein sequences in FASTA format. It is ok to have more than one "source sequence" per bait. The ID for each sequence should include the bait source and the protein ID, separated by a hyphen. For example:
>Amborella-atpH
MNPLISAASVIAAGLAVGLASIGPGVGQGTAAGQAVEGIARQPEAEGKIRGTLLLSLAFM
>Aneura-atpH
MNPLIPAASVIAAGLAVGLASIGPGIGQGTAAGQAVEGIARQPEAEGKIRGTLLSSPASM
>Amborella-rbcL
MSPKTETKASAGFKAGVKDYRLTYYTPDYETLATDILAAFRVTPQPGVPPEEAGAAVAAE
>Aneura-rbcL
MSPQTETKAGVGFKAGVKDYRLTYYTPEYETKETDILAAFRMTPQPGVPPEEAGAAVAAE
For a full tutorial using a test dataset, check out the Tutorial wiki.
HybPiper is run separately for each sample (single or paired-end sequence reads). When HybPiper generates sequence files from the reads, it does so in a standardized directory hierarchy. Many of the post-processing scripts rely on this directory hierarchy, so do not modify it after running the initial pipeline. It is a good idea to run the pipeline for each sample from the same directory. You will end up with one directory per run of HybPiper, and some of the later scripts take advantage of this predictable directory structure.
To execute the entire pipeline, create a directory containing the paired-end read files.
The script reads_first.py will create a directory based on the fastq filenames (or use the --prefix flag):
Anomodon-rostratus_L0001_R1.fastq ---> Anomodon-rostratus/
The following command will execute the entire pipeline on a pair of Illumina read files, using the baits in the file baits.fasta. The HybPiper scripts should be in a different directory:
python /Users/mehmattski/HybPiper/reads_first.py -r MySpecies_R1.fastq MySpecies_R2.fastq -b baits.fasta
The BLASTx version of the pipeline (default) will map the reads to amino acid bait sequences sequences. Although it is slower than the BWA version, it may have higher specificity. Reads may not align to divergent nucleotide bait sequences, which are required for the BWA version. If you find the recovery efficiency is poor with BWA, you may want to try again with BLASTx.
A wrapper script that:
-
Can check if all dependencies are installed correctly. (
--check-depend) -
Creates sub-directories.
-
Calls downstream analyses
You can tell the script to skip upstream steps (for example: --no-blast) but the script will still assume that the output files of these steps still exist!
This script will call, in order:
-
Blastx (or BWA)
-
distribute_reads_to_targets.py(ordistribute_reads_to_targets_bwa.py) anddistribute_targets.py -
Run SPAdes assembler.
-
exonerate_hits.py
Some program-specific options may be passed at the command line.
For example, the e-value threshold for BLASTX (--evalue, default is 1e-9), the coverage-cutoff level for SPAdes assemblies (--cov_cutoff, default is 8), or the percent-identity threshold for aligning contigs to the reference sequences (--thresh, the default is 55).
Use reads_first.py -h for a full list of customizable parameters
After a BLASTx search of the raw reads against the target sequences, the reads need to be sorted according to the successful hits. This script takes the BLASTx output (tabular) and the raw read files, and distributes the reads into FASTA files ready for assembly.
If there are multiple BLAST results (for example, one for each read direction), concatenate them prior to sorting.
After a BWA search of the raw reads against the target sequences, the reads need to be sorted according to the successful hits. This script takes the bam output (parsed using samtools) and the raw read files, and distributes the reads into FASTA files ready for assembly.
If there are multiple BAM results (for example, one for each read direction), concatenate them prior to sorting.
Given a file containing all of the "baits" for a target enrichment, create separate FASTA files with all copies of that bait. Multiple copies of the same bait can be specified using a "-" delimiter. For example, the hits against the following two baits will be sorted in to the same file:
Anomodon-rbcl
Physcomitrella-rbcl
Given multiple baits, the script will choose the most appropriate 'reference' sequence using the highest cumulative BLAST scores across all hits. If the search was BWA rather than BLAST, it will use the bait with the highest total BWA alignment score for each gene.
Output directories can also be created, one for each target category (the default is to put them all in the current one). The field delimiter may also be changed.
This script generates alignments of SPAdes contigs against the target sequence.
If BLASTx is used, the model is protein2genome
If BWA is used, the model is coding2genome
Contigs are not expected to overlap much. An inital exonerate search is filtered for hits that are above a certain threshold (default is 55, can be changed in reads_first.py with --thresh). Contigs that pass this filter are arranged in order along the alignment. If one contig completely subsumes the range of another contig, the longer contig is saved.
To maximize the chance that Exonerate may find introns, all contigs that pass the previous steps are concatenated into a "supercontig" and the exonerate search is repeated. Once again, unique hits that pass a percent identity threshold and do not overlap with longer hits are saved, and from this the full length CDS and protein sequences are generated.
In the older version of the script ("assemble-first"), the script was used after the query_file_builder is complete. The minimal inputs are the tailored target file and the assembly.
If run immediately after query_file_builder, use the --prefix flag to specify the file names:
EXAMPLE COMMAND LINE:
exonerate_hits.py speciesName/baitfile.FAA speciesName/assembly.fasta --prefix=speciesName
The threshold for accepting an exonerate hit can be adjusted with -t (Default: 55 percent)
Optional utilities after running the pipeline for multiple assemblies:
NOTE: for these utilities to work, the files must be in the same directory hierarchy created by the pipeline. (i.e. species/sequences/FAA/ and species/sequences/FNA/)
HybPiper generates a lot of output files. Most of these can be discarded after the run. This script handles deleting unnecessary files, and can reduce the size of the directory created by HybPiper by 75%.
python cleanup.py hyb_seq_directory
By default the script will delete all the files generated by Spades. Other options may be added in the future.
This script will summarize the recovery of genes from multiple samples. If you have all of these separate runs of the HybPiper in the same directory, create a namelist.txt file that contains a list of all the HybPiper directories for each sample (one per line):
Sample1
Sample2
Sample3
Specify the location of the target file and whether it is amino acid or nucleotide on the command line:
python get_seq_lengths.py baitfile.fasta namelist.txt dna > gene_lengths.txt
The script will output a table to stdout. The first line is a header containing the gene names. The second line contains the length of the reference for each gene. If there are multiple reference sequences for each gene, an average is reported. The remaining lines are the lengths recovered by the HybPiper for each sample, one sample per line (one column per gene). If the gene is missing, a 0 is indicated.
A warning will print to stderr if any sequences are longer than 1.5x the average reference length for that gene.
Species 26S 18S
MeanLength 3252.0 1821.6
funaria 3660 1758
timmia 3057 1821
This script will summarize target enrichment and gene recovery efficiency for a set of samples. The output is a text file with one sample per line and the following statistics:
- Number of reads
- Number of reads on target
- Percent reads on target
- Number of genes with reads
- Number of genes with contigs
- Number of genes with sequences
- Number of genes with sequences > 25% of the target length
- Number of genes with sequences > 50% of the target length
- Number of genes with sequences > 75% of the target length
- Number of genes with sequences > 150% of the target length
- Number of genes with paralog warnings
NOTE: The number of reads and percent reads on target will only be reported if mapping was done using BWA.
Example Command Line
python hybpiper_stats.py test_seq_lengths.txt namelist.txt > test_stats.txt
This script takes the ouput of get_seq_lengths.py and creates a figure to visualize the recovery efficiency.
Unlike the python scripts, you will need to open the R script in a text editor or RStudio and edit a few parameters before running the script within R.
The script requires two R packages: gplots and heatmap.plus
Install these using install.packages before running the script.
You will need to set the name of your input file (the one produced by get_seq_lengths.py) at the top of the script.
The output will look something like this:

Each row shows a sample, and each column is a gene. The amount of shading in each box corresponds to the length of the gene recovered for that sample, relative to the length of the reference (target coding sequence).
In this case, there are a few samples for which few or no genes were recovered (white rows) and a few genes that were not recovered in any sample (white columns).
This script fetches the sequences recovered from the same gene for many samples and generates an unaligned multi-FASTA file for each gene.
This script will get the sequences generated from multiple runs of the HybPiper (reads_first.py). Have all of the runs in the same directory (sequence_dir). It retreives all the gene names from the target file used in the run of the pipeline.
python retrieve_sequences.py baitfile.fasta sequence_dir dna
You must specify whether you want the protein (aa) or nucleotide (dna) sequences.
If you ran intronerate.py on these samples, you can also specify "supercontig" or "intron" to recover those sequences instead.
Will output unaligned fasta files, one per gene, to the current directory.
(Specified by --prefix or generated from the read file names)
- The master target file.
- A BLAST (
prefix.blastx) or BWA database (prefix.bam) is generated - One directory is generated for every gene with BLAST or BWA hits.
- A file
bait_tallies.txtsummarizes which target sources were chosen. -
spades_genelist.txt: List of genes with mapped reads. -
exonerate_genelist.txt: List of genes with assembled contigs. -
genes_with_seqs.txt: List of genes for which a coding sequence was extracted. -
genes_with_paralog_warnings.txt: List of genes which had multiple long-length contigs (putative paralogs).
####If intronerate.py is run
-
intron_stats.txt: List of genes and the number of internal introns recovered.
-
GeneName_interleaved.fasta: All reads that mapped to the target. (The read pairs are interleaved.) - SPAdes output. Final assembly is in
GeneName_contigs.fasta -
GeneName_baits.fasta: Fasta file for reference target chosen by thedistribute_targets.pyscript. - Directory of Exonerate results (with same name as the sample)
-
exonerate_results.fasta-- Results of the initial exonerate search for all contigs. -
supercontig_exonerate.fasta-- Long concatenated contig from final exonerate search. -
exonerate_stats.csv: information on all contigs with exonerate hits against the target sequence -
sequencesdirectory
-
intronerate_raw.gff: raw GFF output of exonerate, contains comments -
interonerate.gff: Modified GFF output used byinteronerate.py
- paralogs directory
-
FNA/GeneName.FNA: In-frame nucleotide sequence -
FAA/GeneName.FAA: Amino acid sequence.
-
intron/GeneName_introns.fasta: intron sequence recovered -
intron/GeneName_supercontig.fasta: gene region sequence (introns + exons)
The major steps of the pipeline include:
-
Blast (or BWA) search of the reads against the target sequences.
-
Distribution of reads into separate directories, one per gene.
-
Assembly of reads for each gene into contigs with SPAdes.
-
Conduct one or more exonerate searches for each contig in the assembly. If multiple contigs match the same protein in non-overlapping sequences, stitch the hits together into a “supercontig”
-
In a subdirectory, generate separate FASTA files containing either the nucleotide (FNA) or amino acid (FAA) sequence for each protein.
In many HybSeq bait designs, care is taken to avoid enrichment for genes with paralogous sequences in the target genomes. However, gene duplication and paleopolyploidy (especially in plants) makes it difficult to completely avoid paralogs. Given enough read depth, it may be possible to identify paralogous sequences with HybPiper.
If SPAdes assembler generates multiple contigs that contain coding sequences represeting 75% of the length of the reference protein, HybPiper will print a warning for that gene. It will also print the names of the contigs in prefix/genename/paralog_warning.txt and will print a list of all genes with paralog warnings to prefix/genes_with_paralog_warnings.txt.
If many of your genes have paralogs, one approach could be to add the paralog coding sequence to your target file as a separate gene, and re-running HybPiper. Reads that have a better mapping to the paralog will be sorted accordingly.
For more in-depth discussion about paralogs, see the examples in the paralog tutorial.