 ##Transcriptome assembly with Oases
##Transcriptome assembly with Oases
All of the scripts you will need to complete this lab as well as the sample dataset will be copied to your Beocat directory as you follow the instructions below. You should type or paste the text in the beige code block into your terminal as you follow along with the instructions below. If you are not used to commandline, practice with real data is one of the best ways to learn.
If you would like a quick primer on basic linux commands try these 10 minute lessons from Software Carpentry http://software-carpentry.org/v4/shell/index.html. To learn to start using Beocat and begin using the terminal go to https://github.com/i5K-KINBRE-script-share/FAQ/blob/master/UsingBeocat.md. Learn how to download files from Beocat at https://github.com/i5K-KINBRE-script-share/FAQ/blob/master/BeocatEditingTransferingFiles.md.
We will be using the script "AssembleT.pl" to organize our working directory and write scripts to clean our reads, assemble our de novo transcriptome for human breast cancer cell lines, and summarize our assembly metrics.
To begin this lab your should read about the software we will be using. Prinseq will be used to clean raw reads. Priseq cleaning is highly customizable. You can see a detailed parameter list by typing "perl /homes/sheltonj/abjc/prinseq-lite-0.20.3/prinseq-lite.pl -h" or by visiting their manual at http://prinseq.sourceforge.net/manual.html. You can read a detailed list of parameter options for Oases transcriptome assembler by typing "/homes/sheltonj/abjc/oases_0.2.08/oases" or visit the Oases manual at https://www.ebi.ac.uk/~zerbino/oases/.
To find out more about the parameters for "AssembleT.pl" run "perl ~/transcriptome-and-genome-assembly/KSU_bioinfo_lab/AssembleT/AssembleT.pl -man" or visit its manual at https://github.com/i5K-KINBRE-script-share/transcriptome-and-genome-assembly/tree/master/KSU_bioinfo_lab/AssembleT/AssembleT_MANUAL.md.
###Step 1: Clone the Git repositories
git clone https://github.com/i5K-KINBRE-script-share/transcriptome-and-genome-assembly
git clone https://github.com/i5K-KINBRE-script-share/read-cleaning-format-conversion
git clone https://github.com/i5K-KINBRE-script-share/genome-annotation-and-comparison
###Step 2: Create project directory and add your input data to it
Make a working directory.
mkdir de_novo_transcriptome
cd de_novo_transcriptome
Create symbolic links to raw RNA reads from the human breast cancer cell lines. Creating a symbolic link rather than copying avoids wasting disk space and protects your raw data from being altered. We are using Illumina paired end reads from four breast cancer cell lines. This data was used in a biological visualization competition that illumina held a few years ago http://blog.expressionplot.com/2011/03/15/idea-challenge-2011-illuminas-data-excellence-award/.
ln -s /homes/bioinfo/pipeline_datasets/AssembleT/* ~/de_novo_transcriptome/
Step 3: Write assembly scripts
Check to see if your fastq headers end in "/1" or "/2" (if they do not you must add the parameter "-c" when you run "AssembleT.pl"
head ~/de_novo_transcriptome/*_1.fastq
Your output will look similar to the output below for the sample data. Because these reads end in "/1" or "/2" we will not add "-c" when we call "AssembleT.pl".
==> /homes/bioinfo/de_novo_transcriptome/BT20_paired-end_RNA-seq_subsampled_1.fastq <==
@HWUSI-EAS1794_0001_FC61KOJ:4:30:19389:13787#0/1
GCGGCCCGGCCCCGGCCCCCTGCTCGTTGGCTGTGGCAGGGCCGCCGTGG
+
HHHHHHHGEHHHHDHDHHHHBDGBBC@CAC?8C><AAAACD>DDB?####
@HWUSI-EAS1794_0001_FC61KOJ:4:57:10821:2162#0/1
CAGATATCGAAGATGAAGACTTAAAGTTAGAGCTGCGACGACTACGAGAT
+
IIHIIIIIIHHIHIIIIIEIIIIIIIIIIIHHII@IHIIIIHHEIIHIID
@HWUSI-EAS1794_0001_FC61KOJ:4:75:5014:13576#0/1
CTCAGCCACCAGCAGCGGCACCCCCATCTGCAGTTGGCTCTTCTGCTGCT
Call "AssembleT.pl". Our reads are only 50 bp long so we are setting our minimum read length to 35 bp. Generally you want to keep this length ~10 bp shorter than our read length. We would also raise the longest kmer value "-l" to ~61 if our reads were 100bp.
perl ~/transcriptome-and-genome-assembly/KSU_bioinfo_lab/AssembleT/AssembleT.pl -r cell_line_reads_assembly.txt -p cell_line -s 25 -l 39 -i 2 -n 35 -m 33
###Step 4: Run prinseq and the assembly scripts
Clean raw reads. When these jobs are complete go to next step. Test completion by typing "status" in a Beocat session. Download the ".gd" files in the "~/de_novo_transcriptome/cell_line__prinseq" directory and upload them to http://edwards.sdsu.edu/cgi-bin/prinseq/prinseq.cgi?report=1 to evaluate read quality pre and post cleaning.
bash ~/de_novo_transcriptome/cell_line_qsubs/cell_line_qsubs_clean.sh
Concatenate cleaned reads and shuffle sequences for Oases
bash ~/de_novo_transcriptome/cell_line_scripts/cat_reads.sh
Assemble single kmer transcriptomes. When these jobs are complete go to next step. Test completion by typing "status" in a Beocat session.
bash ~/de_novo_transcriptome/cell_line_qsubs/cell_line_qsubs_singlek.sh
Merge single kmer transcriptomes. When these jobs are complete go to next step. Test completion by typing "status" in a Beocat session.
bash ~/de_novo_transcriptome/cell_line_qsubs/cell_line_qsubs_merge.sh
Cluster merged assembly with CDH. Putative transcripts that share 80% identity over 80% of the length are clustered and the longest transcript is printed in the clustered fasta file. This step will also generate assembly metrics and summarize the cleaning step results.
bash ~/de_novo_transcriptome/cell_line_qsubs/cell_line_qsubs_cluster_and_qc.sh
###Why so many assemblies?
The authors of the Oases-M publication compare various single k-mer assemblies and a merged multiple k-mer assembly for transcript completeness, overall sensitivity, etc. These results show the tradeoff between sensitivity of shorter k-mers (producing longer transcripts and/or more of the rare transcripts but more error-ridden transcripts) and specificity of longer k-mers (shorter transcripts and/or fewer of the rare transcripts but more accurate assemblies).
The authors also determine Oases-M is more sensitive and less specific than Trinity. Additionally, they make the case that de novo assemblies may be even lower quality, in comparison to reference-based assemblies, than suggested by the Trinity paper because the gene set used there excluded rare transcripts. Of course increased depth of coverage can increase both assembly sensitivity and specificity.
Here are the basic steps outlined in the paper: I) Run assembler at several k-mer lengths (sensitive (smaller k ) higher quality (larger k)) Velvet- makes unique contigs (analogous to the inchworm from Trinity); the user makes several assemblies with various k-mer lengths Oases- clusters unitigs into "loci" (analogous to chrysalis) and constructs transcript "isoforms" (analogous to butterfly) II) Merge these assemblies
We have added raw read cleaning with Prinseq as a preprocessing step. Basecalling errors in reads increase the complexity of final de bruijn graphs and can erroneously fragment or increase redundancy in the final assembly. We also used Prinseq to deduplicate identical reads because these do not add information to the assembly but take the assembly time and memory to process.
We have also used CDHit to cluster similar putative transcripts because even with the Oases merge step de novo assemblies tend to be highly redundant. Clustering means that we can utilize our merged assembly when the goal is to find isoforms or members of a gene family and the clustered assembly to estimate expression more at the gene level.
###Evaluate your results
Gruenheit et al. 2012 used the multi kmer method as well as exploring the effects of other parameters on final transcriptome assemblies. Using the complex example of autopolyploids the authors of this paper use the parameters k-mer length and coverage cut-off to address a topic that was touched on in Schulz et al 2012. In Gruenheit et al. 2012 both k-mer size and coverage cut-off were manipulated. The authors estimate they assembled 3,171 more transcripts than would have been produced with a single optimized assembly and state.
Below you see the assembly N50 for single kmer assemblies with the same coverage cutoff value. The reads used in this study were 75 bp. The reads used in the Oases-M paper were based on 45bp reads. The reads you used were 50bp.
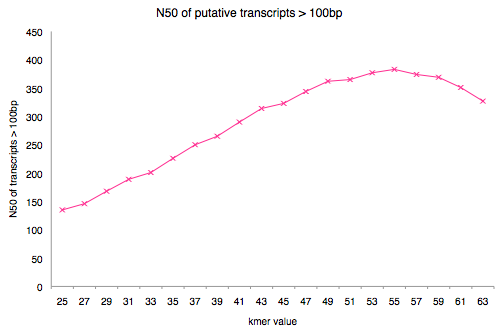
Plot your N50 values from the ~/de_novo_transcriptome/assembly_metrics.csv file.
-
How does your plot compare?
-
If we consider N50 before and after the value that equals 50% of read length how do they compare?
-
Are your results more similar to the OasesM paper or the Gruenheit et al. 2012 paper? Based on this, do you think that with increasing read length we can get higher quality final assemblies?